- Адренолейкодистрофия

Содержание
- 2. Лейкодистрофии Группа заболеваний, характеризующихся диффузной дегенерацией белого вещества головного и спинного мозга, в основе которых лежит
- 3. Х-СЦЕПЛЕННАЯ АДРЕНОЛЕЙКОДИСТРОФИЯ (болезнь Шильдера, суданофильная лейкодистрофия с гиперпигментацией кожных покровов и атрофией надпочечников, диффузный периаксиальный энцефалит,
- 4. Пероксисомные болезни обширная группа наследственных прогрессирующих заболеваний, возникающих в результате нарушения функций пероксисом Пероксисомы присутствуют во
- 5. Этиология и патогенез Х-сцепленное рецессивное заболевание, обусловленое мутациями гена АВСD1, кодирующим трансмембранный белок пероксисом ALDP, что
- 6. Классификация детская церебральная форма ювенильная церебральная форма взрослая церебральная форма адреномиелоневропатия (25%) изолированная надпочечниковая недостаточность асимптомная
- 7. Клинические проявления адренолейкодистрофии Сочетание надпочечниковой недостаточности и поражения нервной системы: бронзовый цвет кожный покровов, подострое развитие
- 8. Церебральные формы Возраст дебюта детской церебральной формы - 7,2±1,7 лет. В 86% случаев неврологические и психические
- 9. Церебральные формы Юношеская форма манифестирует в возрасте 10–21 лет, по клиническим проявлениям сходна с детской церебральной.
- 10. Адреномиелонейропатия Возраст начала - от 12 до 50 лет. Гиперпигментация кожных покровов иногда манифестирует в раннем
- 11. У большинства пациентов с течение времени присоединяются психические нарушения в виде эмоциональных и депрессивных расстройств. Интеллект
- 12. Изолированная надпочечниковая недостаточность Характеризуется симптомами хронической надпочечниковой недостаточности (болезнь Адиссона) - соматические проявления (пигментация кожи, анорексия,
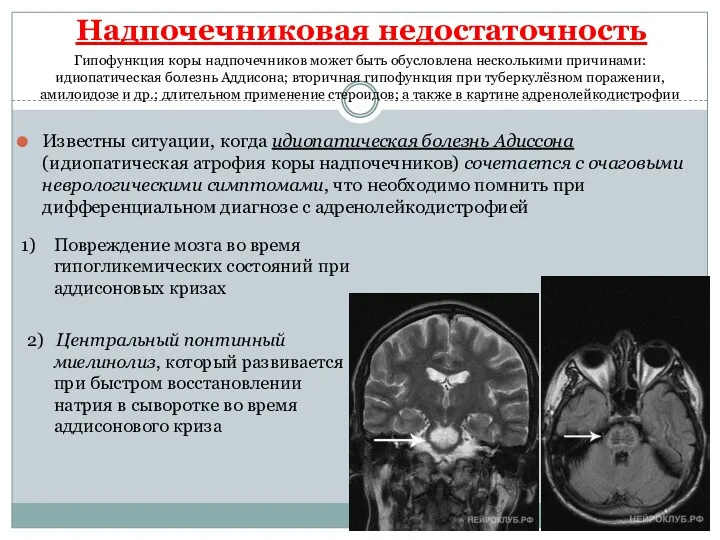
- 13. Надпочечниковая недостаточность Известны ситуации, когда идиопатическая болезнь Адиссона (идиопатическая атрофия коры надпочечников) сочетается с очаговыми неврологическими
- 14. Диагностика Исследование ЦСЖ При церебральных формах Х-АЛД в цереброспинальной жидкости повышается содержание иммуноглобулинов G. Стволовые вызванные
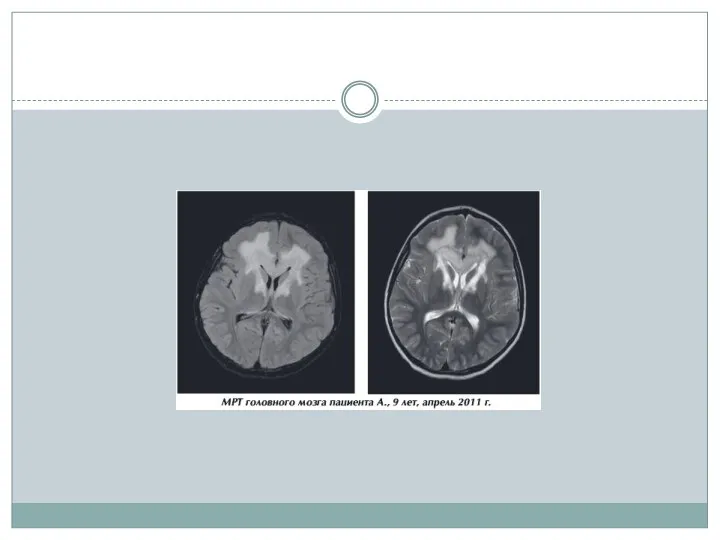
- 15. Нейрорадиологические методы На МРТ у больных с церебральными формами Х-АЛД на начальных стадиях выявляют гиперинтенсивный сигнал
- 18. По мере прогрессирования, на фоне выраженной воспалительной реакции в очагах демиелинизации, нарушения проницаемости ГЭБ, выявляется накопление
- 19. Биохимическая диагностика обнаружение в плазме крови, эритроцитах, лейкоцитах, культуре клеток кожных фибробластов повышенного уровня ОДЦЖК, особенно
- 20. Дифференциальный диагноз Дифференциальный диагноз проводят с: первично-прогрессирующей формой рассеянного склероза семейной спастической параплегией Штрюмпеля, шейной миелопатией
- 21. Семейная спастическая параплегия Штрюмпеля Клиническая картина складывается из медленно прогрессирующей слабости в ногах и спастичности с
- 22. Наследственные спастические параплегии «плюс» Наследственная спастическая параплегия с нарушением зрения Дегенерация сетчатки с атрофией зрительных нервов,
- 23. Первично-прогрессирующая форма РС Начинается в более позднем возрасте (около 40 лет), не наблюдается преобладания женщин. Скорость
- 24. Проявлением многоочагового демиелинизирующего процесса является сочетание центрального паралича с гиперрефлексией, клонусами, патологическими знаками и одновременно выраженной
- 25. Шейная миелопатия Представляет собой серьёзное осложнение цервикального спондилёза или, реже, кальцификации задней продольной связки на шейном
- 26. 1) синдром поперечного поражения с вовлечением коритикоспинальных, спино-таламических трактов и проводников задних столбов спинного мозга с
- 27. Энцефалит при ветряной оспе Страдают преимущественно дети от 2-3 до 6 лет. Поражение нервной системы при
- 28. В результате первичного инфицирования вирусы в 100% случаев пожизненно персистируют в нейронах спинальных ганглиев или ЧМН.
- 29. Глобоидно-клеточная лейкодистрофия Краббе — Бенеке связана с недостаточностью фермента галактоцереброзидазы, приводящим к накоплению в участках демиелинизации
- 30. Подострый склерозирующий лейкоэнцефалит Этиология - персистирующая вирусная инфекция (коревая, энтеровирусная, вирус клещевого энцефалита и др.) Заболевают
- 31. Течение неуклонно прогрессирующее и всегда заканчивается летально. Длительность заболевания от 6 мес до 2-3 лет. Смерть
- 32. НИЖНИЙ СПАСТИЧЕСКИЙ ПАРАПАРЕЗ Нижний спастический парапарез развивается при двустороннем поражении верхних моторных нейронов (в области парацентральных
- 33. Диагностические исследования при нижнем спастическом парапарезе 1. МРТ головного мозга, позвоночника и кранио-вертебрального перехода; 2. Миелография;
- 34. Основные причины нижнего спастического парапареза: А. Компрессионные поражения 1. Экстрамедуллярные и интрамедуллярные опухоли спинного мозга 2.
- 35. Основные причины нижнего спастического парапареза: D. Сосудистые болезни 1. Окклюзия передней спинальной артерии 2. Эпидуральная и
- 36. Другие причины нижнего спастического парапареза Первичный боковой склероз редкий вариант болезни мотонейрона с преимущественным поражением верхнего
- 37. Лечение 1) Диетотерапия в сочетании с применением масла Лоренцо 2) Медикаментозная коррекция надпочечниковой недостаточности 3) Симптоматическая
- 38. Масло Лоренцо (Lorenzo’s oil) - смесь мононенасыщеных жирных кислот - эруковой (С22-1) и олеиновой в соотношении
- 39. Всем пациентам с надпочечниковой недостаточностью показано назначение стероидной терапии под контролем уровня гормонов (АКТГ, кортизол), электролитов
- 40. Трансплантация костного мозга — основной метод лечения Х-сцепленной адренолейкодистрофии в настоящее время Лечение 4) Трансплантация гемопоэтических
- 41. Трансплантация костного мозга Для отбора пациентов на проведение ТГСК должны быть проведено тщательное неврологическое обследование, нейропсихологическое
- 42. В будущем планируется проведение генотерапии с применением лентивирусного вектора, что будет являться альтернативным методом трансплантации гемопоэтических
- 43. Диспансерное наблюдение за пациентами с Х-АЛД
- 44. Диспансерное наблюдение за пациентами с Х-АЛД Диспансерное наблюдение за пациентами мужского пола с Х-АЛД является очень
- 45. Профилактика Пренатальная диагностика Х-АЛД проводится с использованием биохимических методов (определение уровня ОДЦЖК в плазме крови)и методов
- 46. Прогноз При адреномиелонейропатии Продолжительность жизни, как правило, не сокращается (Moser et al.,1992) При церебральных формах -
- 47. БЛАГОДАРЮ ЗА ВНИМАНИЕ!!!
- 50. Скачать презентацию

Лейкодистрофии
Группа заболеваний, характеризующихся диффузной дегенерацией белого вещества головного и спинного мозга,
Лейкодистрофии
Группа заболеваний, характеризующихся диффузной дегенерацией белого вещества головного и спинного мозга,

Х-СЦЕПЛЕННАЯ АДРЕНОЛЕЙКОДИСТРОФИЯ
(болезнь Шильдера, суданофильная лейкодистрофия с гиперпигментацией кожных покровов и атрофией
Х-СЦЕПЛЕННАЯ АДРЕНОЛЕЙКОДИСТРОФИЯ
(болезнь Шильдера, суданофильная лейкодистрофия с гиперпигментацией кожных покровов и атрофией

Пероксисомные болезни
обширная группа наследственных прогрессирующих заболеваний, возникающих в результате нарушения функций
Пероксисомные болезни
обширная группа наследственных прогрессирующих заболеваний, возникающих в результате нарушения функций
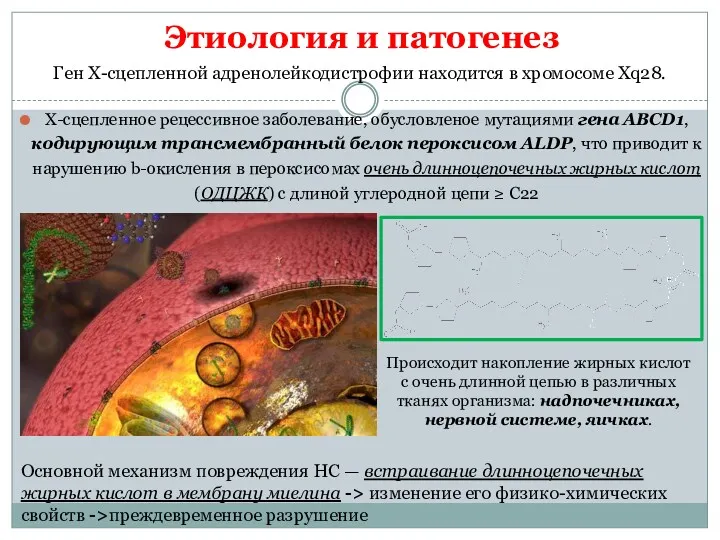
Этиология и патогенез
Х-сцепленное рецессивное заболевание, обусловленое мутациями гена АВСD1, кодирующим трансмембранный
Этиология и патогенез
Х-сцепленное рецессивное заболевание, обусловленое мутациями гена АВСD1, кодирующим трансмембранный

Классификация
детская церебральная форма
ювенильная церебральная форма
взрослая церебральная форма
адреномиелоневропатия (25%)
изолированная надпочечниковая недостаточность
Классификация
детская церебральная форма
ювенильная церебральная форма
взрослая церебральная форма
адреномиелоневропатия (25%)
изолированная надпочечниковая недостаточность
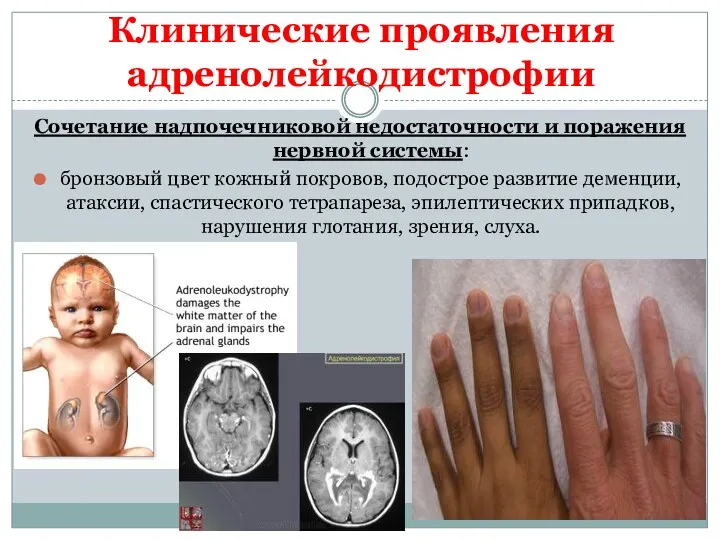
Клинические проявления
адренолейкодистрофии
Сочетание надпочечниковой недостаточности и поражения нервной системы:
бронзовый цвет кожный
Клинические проявления
адренолейкодистрофии
Сочетание надпочечниковой недостаточности и поражения нервной системы:
бронзовый цвет кожный

Церебральные формы
Возраст дебюта детской церебральной формы - 7,2±1,7 лет.
В 86%
Церебральные формы
Возраст дебюта детской церебральной формы - 7,2±1,7 лет.
В 86%

Церебральные формы
Юношеская форма манифестирует в возрасте 10–21 лет, по клиническим проявлениям
Церебральные формы
Юношеская форма манифестирует в возрасте 10–21 лет, по клиническим проявлениям

Адреномиелонейропатия
Возраст начала - от 12 до 50 лет.
Гиперпигментация кожных покровов
Адреномиелонейропатия
Возраст начала - от 12 до 50 лет.
Гиперпигментация кожных покровов
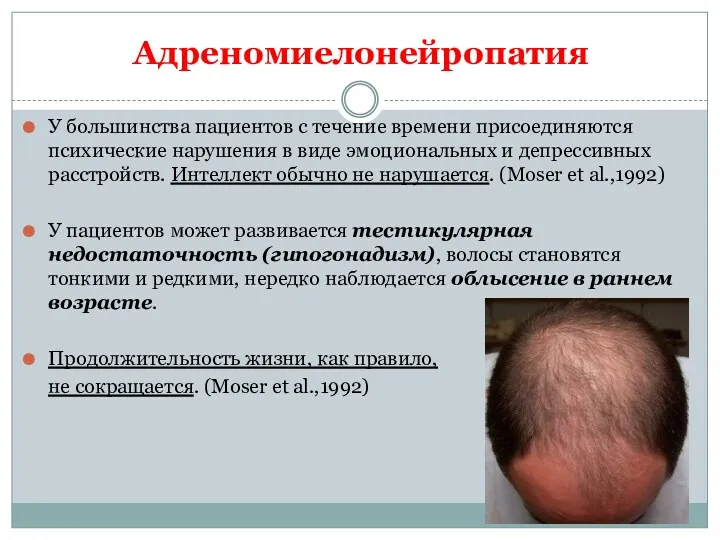
У большинства пациентов с течение времени присоединяются психические нарушения в виде
У большинства пациентов с течение времени присоединяются психические нарушения в виде
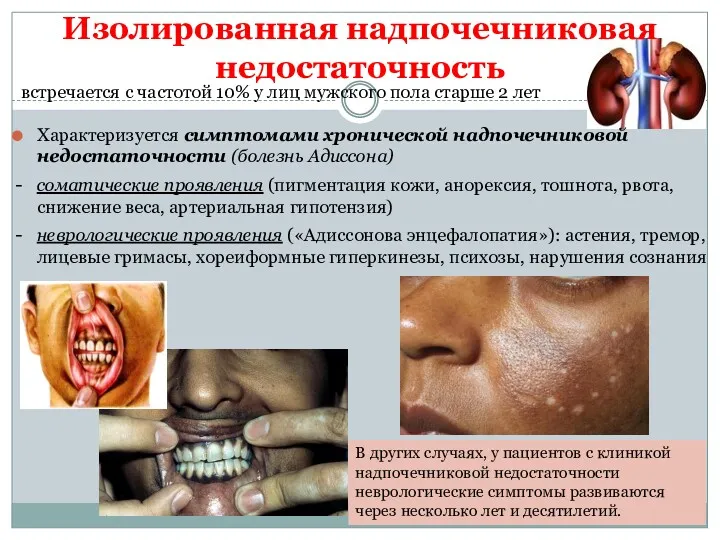
Изолированная надпочечниковая недостаточность
Характеризуется симптомами хронической надпочечниковой недостаточности (болезнь Адиссона)
- соматические проявления
Изолированная надпочечниковая недостаточность
Характеризуется симптомами хронической надпочечниковой недостаточности (болезнь Адиссона)
- соматические проявления
Надпочечниковая недостаточность
Известны ситуации, когда идиопатическая болезнь Адиссона (идиопатическая атрофия коры надпочечников)
Надпочечниковая недостаточность
Известны ситуации, когда идиопатическая болезнь Адиссона (идиопатическая атрофия коры надпочечников)
Диагностика
Исследование ЦСЖ
При церебральных формах Х-АЛД в цереброспинальной жидкости повышается содержание иммуноглобулинов
Диагностика
Исследование ЦСЖ
При церебральных формах Х-АЛД в цереброспинальной жидкости повышается содержание иммуноглобулинов

Нейрорадиологические методы
На МРТ у больных с церебральными формами Х-АЛД на
На МРТ у больных с церебральными формами Х-АЛД на
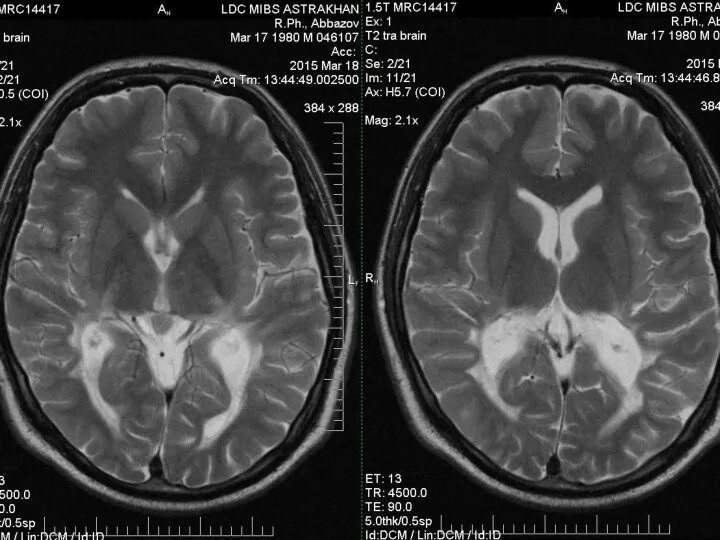
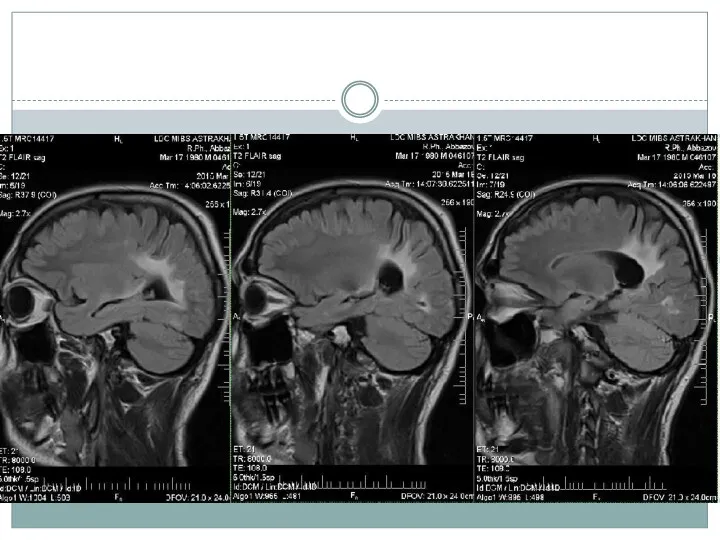

По мере прогрессирования, на фоне выраженной воспалительной реакции в очагах демиелинизации,
По мере прогрессирования, на фоне выраженной воспалительной реакции в очагах демиелинизации,

Биохимическая диагностика
обнаружение в плазме крови, эритроцитах, лейкоцитах, культуре клеток кожных
Биохимическая диагностика
обнаружение в плазме крови, эритроцитах, лейкоцитах, культуре клеток кожных

Дифференциальный диагноз
Дифференциальный диагноз проводят с:
первично-прогрессирующей формой рассеянного склероза
семейной спастической параплегией
Дифференциальный диагноз
Дифференциальный диагноз проводят с:
первично-прогрессирующей формой рассеянного склероза
семейной спастической параплегией

Семейная спастическая параплегия Штрюмпеля
Клиническая картина складывается из медленно прогрессирующей слабости в
Семейная спастическая параплегия Штрюмпеля
Клиническая картина складывается из медленно прогрессирующей слабости в

Наследственные спастические параплегии «плюс»
Наследственная спастическая параплегия с нарушением зрения
Дегенерация сетчатки
Наследственные спастические параплегии «плюс»
Наследственная спастическая параплегия с нарушением зрения
Дегенерация сетчатки
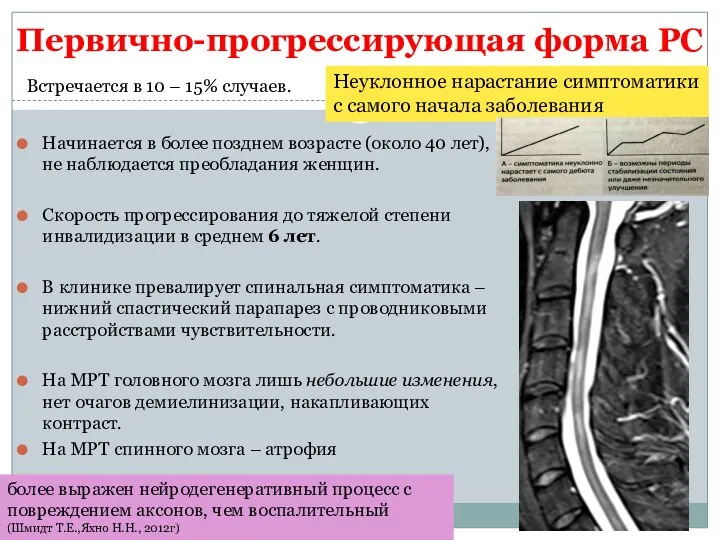
Первично-прогрессирующая форма РС
Начинается в более позднем возрасте (около 40 лет), не
Первично-прогрессирующая форма РС
Начинается в более позднем возрасте (около 40 лет), не
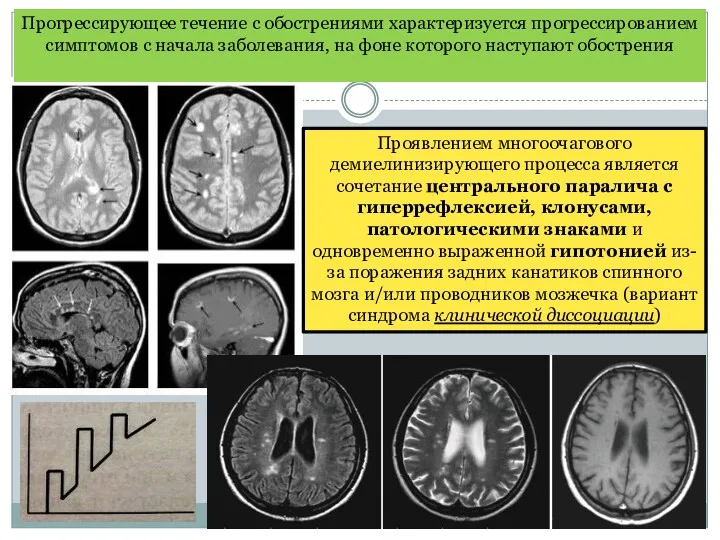
Проявлением многоочагового демиелинизирующего процесса является сочетание центрального паралича с гиперрефлексией, клонусами,
Проявлением многоочагового демиелинизирующего процесса является сочетание центрального паралича с гиперрефлексией, клонусами,
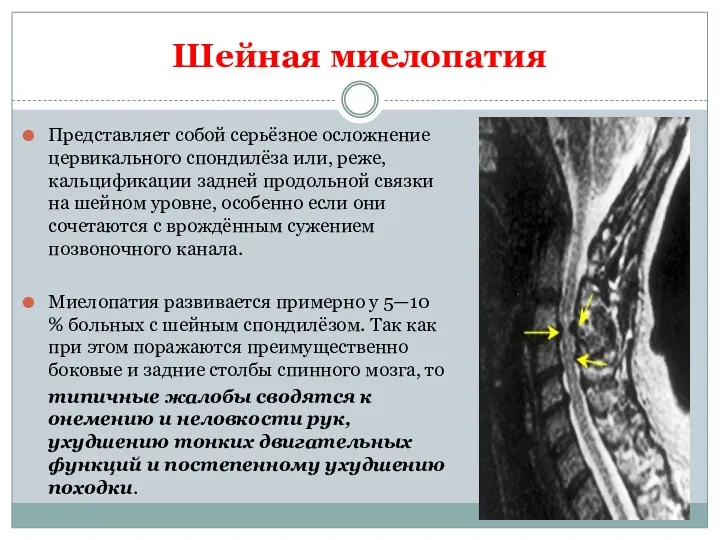
Шейная миелопатия
Представляет собой серьёзное осложнение цервикального спондилёза или, реже, кальцификации задней
Шейная миелопатия
Представляет собой серьёзное осложнение цервикального спондилёза или, реже, кальцификации задней

1) синдром поперечного поражения с вовлечением коритикоспинальных, спино-таламических трактов и проводников
Энцефалит при ветряной оспе
Страдают преимущественно дети от 2-3 до 6 лет.
Энцефалит при ветряной оспе
Страдают преимущественно дети от 2-3 до 6 лет.

В результате первичного инфицирования вирусы в 100% случаев пожизненно персистируют в
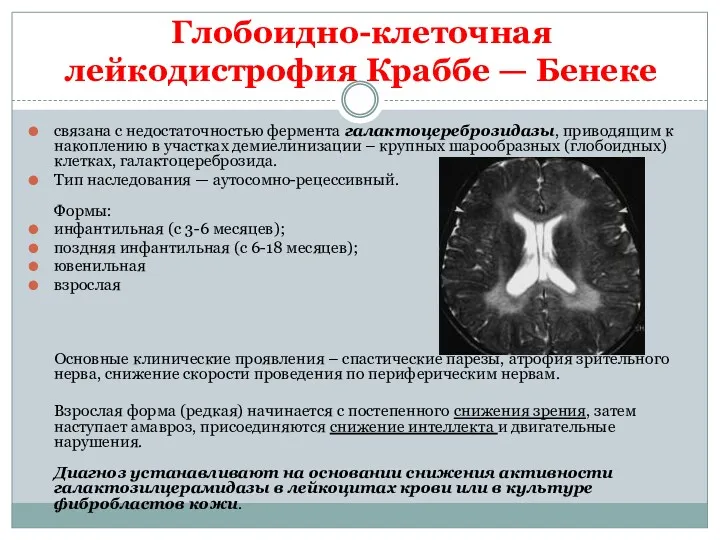
Глобоидно-клеточная лейкодистрофия Краббе — Бенеке
связана с недостаточностью фермента галактоцереброзидазы, приводящим к
Глобоидно-клеточная лейкодистрофия Краббе — Бенеке
связана с недостаточностью фермента галактоцереброзидазы, приводящим к

Подострый склерозирующий лейкоэнцефалит
Этиология - персистирующая вирусная инфекция (коревая, энтеровирусная, вирус клещевого
Подострый склерозирующий лейкоэнцефалит
Этиология - персистирующая вирусная инфекция (коревая, энтеровирусная, вирус клещевого
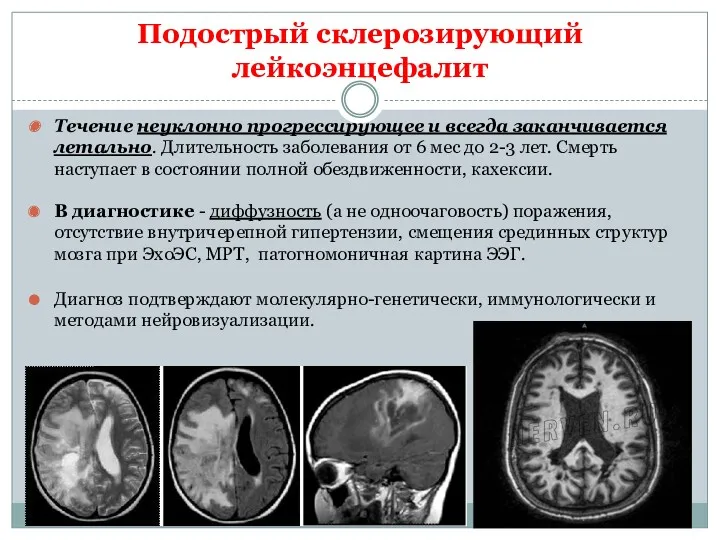
Течение неуклонно прогрессирующее и всегда заканчивается летально. Длительность заболевания от 6
Течение неуклонно прогрессирующее и всегда заканчивается летально. Длительность заболевания от 6

НИЖНИЙ СПАСТИЧЕСКИЙ ПАРАПАРЕЗ
Нижний спастический парапарез развивается при
двустороннем поражении верхних моторных
НИЖНИЙ СПАСТИЧЕСКИЙ ПАРАПАРЕЗ
Нижний спастический парапарез развивается при
двустороннем поражении верхних моторных

Диагностические исследования
при нижнем спастическом парапарезе
1. МРТ головного мозга, позвоночника и кранио-вертебрального
Диагностические исследования
при нижнем спастическом парапарезе
1. МРТ головного мозга, позвоночника и кранио-вертебрального

Основные причины нижнего спастического парапареза:
А. Компрессионные поражения
1. Экстрамедуллярные и интрамедуллярные опухоли
Основные причины нижнего спастического парапареза:
А. Компрессионные поражения 1. Экстрамедуллярные и интрамедуллярные опухоли

Основные причины нижнего спастического парапареза:
D. Сосудистые болезни
1. Окклюзия передней спинальной артерии
2.
Основные причины нижнего спастического парапареза:
D. Сосудистые болезни 1. Окклюзия передней спинальной артерии 2.

Другие причины нижнего спастического парапареза
Первичный боковой склероз редкий вариант болезни
Другие причины нижнего спастического парапареза
Первичный боковой склероз редкий вариант болезни

Лечение
1) Диетотерапия в сочетании с применением масла Лоренцо
2) Медикаментозная коррекция
Лечение
1) Диетотерапия в сочетании с применением масла Лоренцо
2) Медикаментозная коррекция

Масло Лоренцо (Lorenzo’s oil) - смесь мононенасыщеных жирных кислот - эруковой
Масло Лоренцо (Lorenzo’s oil) - смесь мононенасыщеных жирных кислот - эруковой

Всем пациентам с надпочечниковой недостаточностью показано назначение стероидной терапии под контролем
Всем пациентам с надпочечниковой недостаточностью показано назначение стероидной терапии под контролем
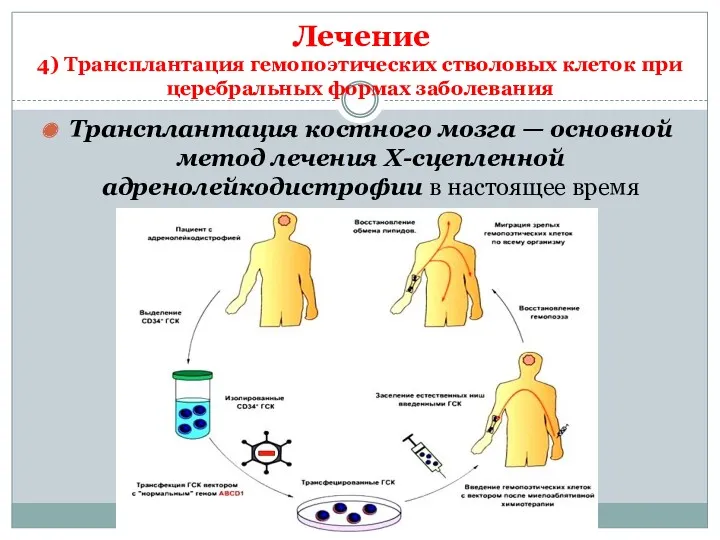
Трансплантация костного мозга — основной метод лечения Х-сцепленной адренолейкодистрофии в настоящее
Трансплантация костного мозга — основной метод лечения Х-сцепленной адренолейкодистрофии в настоящее

Трансплантация костного мозга
Для отбора пациентов на проведение ТГСК должны быть проведено
Трансплантация костного мозга
Для отбора пациентов на проведение ТГСК должны быть проведено

В будущем планируется проведение генотерапии с применением лентивирусного вектора, что будет
В будущем планируется проведение генотерапии с применением лентивирусного вектора, что будет
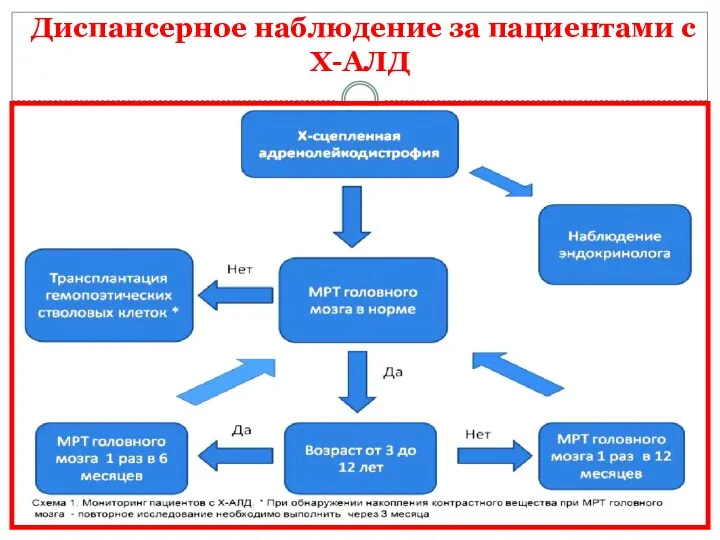
Диспансерное наблюдение за пациентами с Х-АЛД
Диспансерное наблюдение за пациентами с Х-АЛД

Диспансерное наблюдение за пациентами с Х-АЛД
Диспансерное наблюдение за пациентами мужского пола
Диспансерное наблюдение за пациентами с Х-АЛД
Диспансерное наблюдение за пациентами мужского пола

Профилактика
Пренатальная диагностика Х-АЛД проводится с использованием биохимических методов (определение уровня ОДЦЖК
Профилактика
Пренатальная диагностика Х-АЛД проводится с использованием биохимических методов (определение уровня ОДЦЖК

Прогноз
При адреномиелонейропатии Продолжительность жизни, как правило, не сокращается (Moser et
Прогноз
При адреномиелонейропатии Продолжительность жизни, как правило, не сокращается (Moser et

БЛАГОДАРЮ
ЗА
ВНИМАНИЕ!!!
БЛАГОДАРЮ
ЗА
ВНИМАНИЕ!!!
 Vibrio cholerae
Vibrio cholerae Застосування турнікетів для кінцівок
Застосування турнікетів для кінцівок Топографическая анатомия головы
Топографическая анатомия головы Система крови
Система крови Определение функционального состояния организма с использованием расчетных методов исследования
Определение функционального состояния организма с использованием расчетных методов исследования Патофизиология сердечно-сосудистой системы
Патофизиология сердечно-сосудистой системы Нарушения водно-электролитного обмена
Нарушения водно-электролитного обмена Ранняя помощь в условиях реабилитационного центра
Ранняя помощь в условиях реабилитационного центра Особенности травматизма детского возраста
Особенности травматизма детского возраста Варикоцеле. Схематическое изображение и фото варикоцеле
Варикоцеле. Схематическое изображение и фото варикоцеле Острая лучевая болезнь
Острая лучевая болезнь Онкогендер мен олардың ісіктену процесіндегі рөлі туралы қазіргі көзқарас
Онкогендер мен олардың ісіктену процесіндегі рөлі туралы қазіргі көзқарас Медициналық заттар мен инструменттерді залалсыздандыру түрлері мен әдістері
Медициналық заттар мен инструменттерді залалсыздандыру түрлері мен әдістері Время и пространство в лечебной педагогике
Время и пространство в лечебной педагогике Туберкулёз. Пути заражения
Туберкулёз. Пути заражения Муковисцидоз
Муковисцидоз Кальций-Д3 Никомед Форте
Кальций-Д3 Никомед Форте Гигиенические требования к планировке оборудования, содержание детских и подростковых учреждений
Гигиенические требования к планировке оборудования, содержание детских и подростковых учреждений Гнездная алопеция
Гнездная алопеция Интоксикация животных лекарственными средствами
Интоксикация животных лекарственными средствами Регуляция сосудистого тонуса у критических больных
Регуляция сосудистого тонуса у критических больных Медико-генетическое консультирование
Медико-генетическое консультирование Факторы риска неинфекционных заболеваний
Факторы риска неинфекционных заболеваний Патологоанатомическая диагностика болезней сердца у кошек в ветеринарной клинике
Патологоанатомическая диагностика болезней сердца у кошек в ветеринарной клинике Меню 3-7 лет(сад)
Меню 3-7 лет(сад) Естің бұзылыстары. Нарушения памяти
Естің бұзылыстары. Нарушения памяти Ινσουλινη. Παραγοπντες που ρυθμιζουν την εκκριση της ινσουλινης
Ινσουλινη. Παραγοπντες που ρυθμιζουν την εκκριση της ινσουλινης Жылан уы. Бал ара тіршілігінің өнімдері
Жылан уы. Бал ара тіршілігінің өнімдері