- Биохимическая генетика

Содержание
- 2. Болезни обмена аминокислот у детей
- 3. Суммарная частота среди новорожденных 1:2 000 – 1:5 000, среди детей с нарушением развития 1:3 –
- 4. Фенилкетонурия Фенилкетонурия (phenilketonuria; фенилаланин + кетоны + греч. uron моча; син.: оксифенилкетонурия, олигофрения фенилпировиноградная, Феллинга болезнь,
- 5. Биохимические блоки при фенилкетонурии, алкаптонурии, врожденном гипотиреозе и глазо - кожном альбинизме.
- 6. Больной с фенилкетонурией
- 7. Лечение классической ФКУ: диетотерапия ограничение приема белка (до 2 г/кг) и фенилаланина (10-50 мг/кг) за счет
- 8. Лечение классической ФКУ Эффективность лечения зависит от сроков его начала (до 8 недель жизни) и уровня
- 9. Последствия неадекватной терапии Низкорослость Переломы конечностей вследствие остеопороза Психопатологические расстройства: снижение познавательных способностей эмоционально-волевые нарушения девиантное
- 10. Гетерогенность ФКУ Классическая ФКУ Гиперфенилаланинемия новорожденных вследствие транзиторной незрелости печеночных ферментов. Атипичная ФКУ – дефицит ферментов
- 11. Фенилкетонурия (атипичная) Тип наследования – аутосомно-рецессивный Частота 1:30 000 новорожденных Локализация гена 4p15.31 Дефицит ферментов синтеза
- 12. Фенилкетонурия (атипичная) Клинические признаки – мышечная гипотония, задержка психомоторного развития, судороги, тетрапарез, экзематозные изменения кожи, необычный
- 13. Материнская фенилкетонурия (эмбриофетопатия) Сроки манифестации – с рождения Клинические признаки – умственная отсталость (75-90 %) микроцефалия
- 15. Биохимические блоки при фенилкетонурии, алкаптонурии, врожденном гипотиреозе и глазо - кожном альбинизме.
- 16. Глазо-кожный альбинизм у афроамериканского ребенка
- 17. Гомоцистинурия
- 18. Характеристика некоторых аминоацидопатий
- 19. Нарушения обмена углеводов
- 20. Галактоземия Галактоземия (galactosaemia; галактоза + греч. haima кровь; син. олигофрения галактоземическая) - наследственная болезнь, обусловленная нарушением
- 21. Симптомы галактоземии
- 22. Галактоземия
- 23. Галактоза
- 25. Метаболизм галактозы Галактоза внеш. Транспортер галактозы Галактокиназа GALK Галактозо-1-фосфат UDP-галактоза Глюкозо-1-фосфат Галактоза внутр. UDP-глюкоза UDP-галактозо-4-эпимераза (GALE)
- 26. Метаболизм галактозы при галактоземии Галактоза внеш. Транспортер галактозы Галактокиназа GALK Галактозо-1-фосфат UDP-галактоза Глюкозо-1-фосфат Галактоза внутр. UDP-глюкоза
- 27. Характеристика дефектов обмена углеводов
- 28. Нарушения обмена жиров
- 29. Множественные ксантомы при семейной гиперхолестеринемии
- 34. Схема строения липопротеина плазмы крови Интегральный апобелок Свободный холестерол Периферический апобелок Фосфолипиды Триацилглицерол Эфир холестерина Ядро,
- 35. Стадии биосинтеза холестерина и метаболизма липопротеинов никой плотности, указывающие на различные причины семейной гиперхолестеринемии
- 36. Болезни обмена микроэлементов.
- 37. Болезнь Вильсона (гепатолентикулярная дегенерация) Кольцо Кайзера-Флейшера. Отложения меди по краю радужной оболочки глаза. Для нее характерны
- 38. Скрининг на частые наследственные болезни
- 39. Общие характеристики скрининга Массовый безотборный характер обследования Профилактическая направленность Двухэтапность диагностики
- 40. Критерии отбора заболеваний для скрининга Болезнь без лечения существенно снижает жизнеспособность, ведет к инвалидности. Имеются биохимические
- 41. Критерии отбора заболеваний для скрининга Существуют эффективные методы лечения болезни. Частота заболевания 1:10 000 и выше.
- 42. Требования к методам скрининга Экономичность. Методы диагностики болезни должны быть простыми и дешевыми Чувтвительность и специфичность
- 43. Этапы скрининга Взятие биологического материала у новорожденных и доставка в лабораторию Лабораторная просеивающая диагностика Уточняющая диагностика
- 44. Нозологические формы, подлежащие скринингу в Российской Федерации Фенилкетонурия Галактоземия Врожденный гипотиреоз Муковисцидоз Врожденная гиперплазия коры надпочечников
- 45. Скрининг на ФКУ Определение уровня фенилаланина в крови, взятой у новорожденных на 4 – 5-й день
- 46. Скрининг нагалактоземию Определение уровня галактозы в крови, взятой у новорожденных на 4 – 5-й день жизни
- 47. Врожденный гипотиреоз Гипотиреоз (hypothyreosis; гипо- + анат. glandula thyreoidea щитовидная железа + -оз; син.: гипотиреоидизм) -
- 50. Муковисцидоз Муковисцидоз (mucoviscidosis; мука- + лат. viscidus липкий + -оз; син.: диспория энтеробронхопанкреатическая, панкреофиброз, стеаторея панкреатическая
- 52. Адреногенитальный синдром АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ (врожденная дисфункция коры надпочечников, врожденная гиперплазия коры надпочечников) - группа наследственных болезней,
- 54. Скачать презентацию

Болезни обмена аминокислот у детей
Болезни обмена аминокислот у детей

Суммарная частота
среди новорожденных 1:2 000 –
1:5 000, среди детей с
Суммарная частота среди новорожденных 1:2 000 – 1:5 000, среди детей с

Фенилкетонурия
Фенилкетонурия (phenilketonuria; фенилаланин + кетоны + греч. uron моча; син.: оксифенилкетонурия,
Фенилкетонурия
Фенилкетонурия (phenilketonuria; фенилаланин + кетоны + греч. uron моча; син.: оксифенилкетонурия,
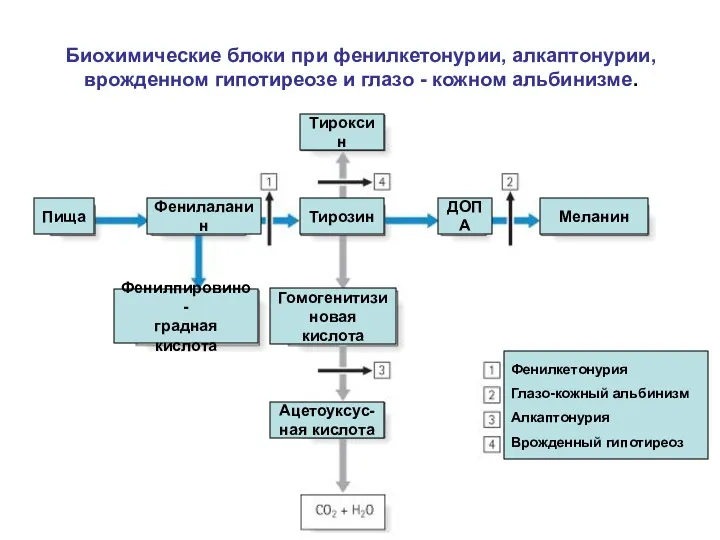
Биохимические блоки при фенилкетонурии, алкаптонурии, врожденном гипотиреозе и глазо - кожном
Биохимические блоки при фенилкетонурии, алкаптонурии, врожденном гипотиреозе и глазо - кожном

Больной с фенилкетонурией
Больной с фенилкетонурией

Лечение классической ФКУ: диетотерапия
ограничение приема белка (до 2 г/кг) и фенилаланина
Лечение классической ФКУ: диетотерапия
ограничение приема белка (до 2 г/кг) и фенилаланина

Лечение классической ФКУ
Эффективность лечения зависит от сроков его начала (до 8
Лечение классической ФКУ
Эффективность лечения зависит от сроков его начала (до 8

Последствия неадекватной терапии
Низкорослость
Переломы конечностей вследствие остеопороза
Психопатологические расстройства:
снижение познавательных способностей
эмоционально-волевые
Последствия неадекватной терапии
Низкорослость
Переломы конечностей вследствие остеопороза
Психопатологические расстройства:
снижение познавательных способностей
эмоционально-волевые

Гетерогенность ФКУ
Классическая ФКУ
Гиперфенилаланинемия новорожденных вследствие транзиторной незрелости печеночных ферментов.
Атипичная ФКУ –
Гетерогенность ФКУ
Классическая ФКУ
Гиперфенилаланинемия новорожденных вследствие транзиторной незрелости печеночных ферментов.
Атипичная ФКУ –

Фенилкетонурия (атипичная)
Тип наследования – аутосомно-рецессивный
Частота 1:30 000 новорожденных
Локализация гена 4p15.31
Дефицит ферментов
Фенилкетонурия (атипичная)
Тип наследования – аутосомно-рецессивный
Частота 1:30 000 новорожденных
Локализация гена 4p15.31
Дефицит ферментов

Фенилкетонурия (атипичная)
Клинические признаки – мышечная гипотония, задержка психомоторного развития, судороги, тетрапарез,
Фенилкетонурия (атипичная)
Клинические признаки – мышечная гипотония, задержка психомоторного развития, судороги, тетрапарез,

Материнская фенилкетонурия (эмбриофетопатия)
Сроки манифестации – с рождения
Клинические признаки – умственная
Материнская фенилкетонурия (эмбриофетопатия)
Сроки манифестации – с рождения
Клинические признаки – умственная

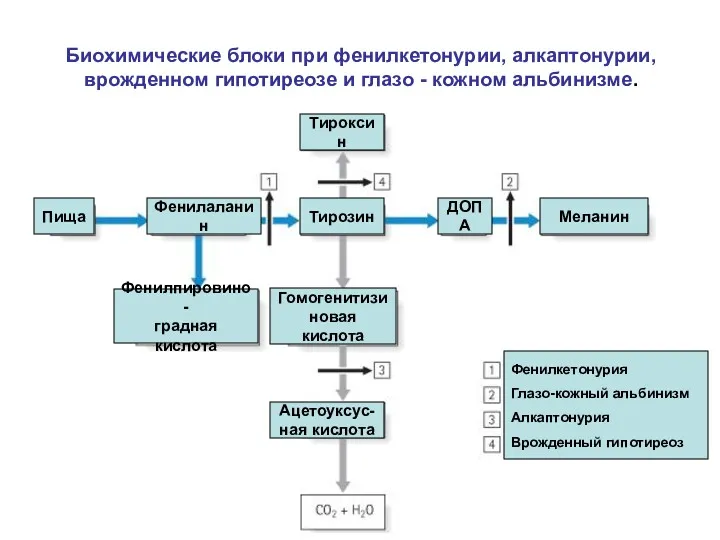
Биохимические блоки при фенилкетонурии, алкаптонурии, врожденном гипотиреозе и глазо - кожном
Биохимические блоки при фенилкетонурии, алкаптонурии, врожденном гипотиреозе и глазо - кожном

Глазо-кожный альбинизм у афроамериканского ребенка
Глазо-кожный альбинизм у афроамериканского ребенка

Гомоцистинурия
Гомоцистинурия
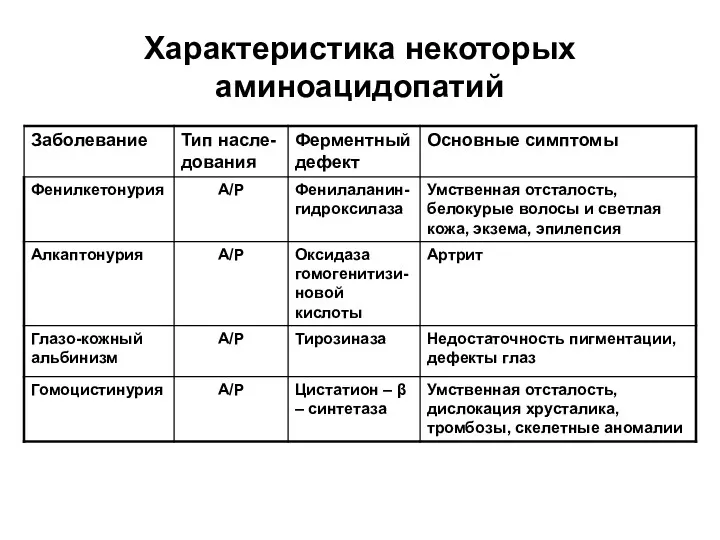
Характеристика некоторых аминоацидопатий
Характеристика некоторых аминоацидопатий

Нарушения обмена углеводов
Нарушения обмена углеводов

Галактоземия
Галактоземия (galactosaemia; галактоза + греч. haima кровь; син. олигофрения галактоземическая) -
Галактоземия
Галактоземия (galactosaemia; галактоза + греч. haima кровь; син. олигофрения галактоземическая) -
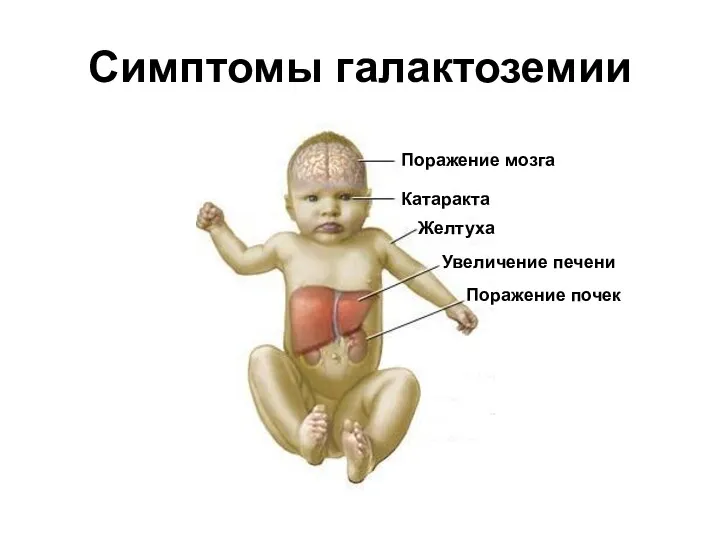
Симптомы галактоземии
Симптомы галактоземии
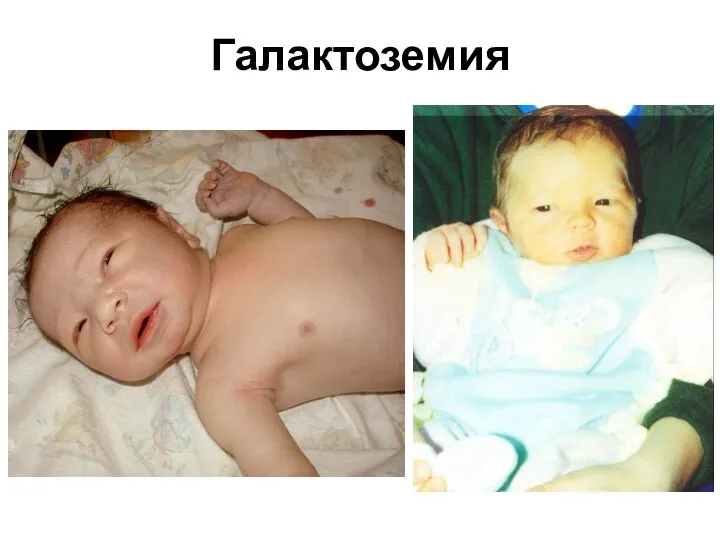
Галактоземия
Галактоземия

Галактоза
Галактоза
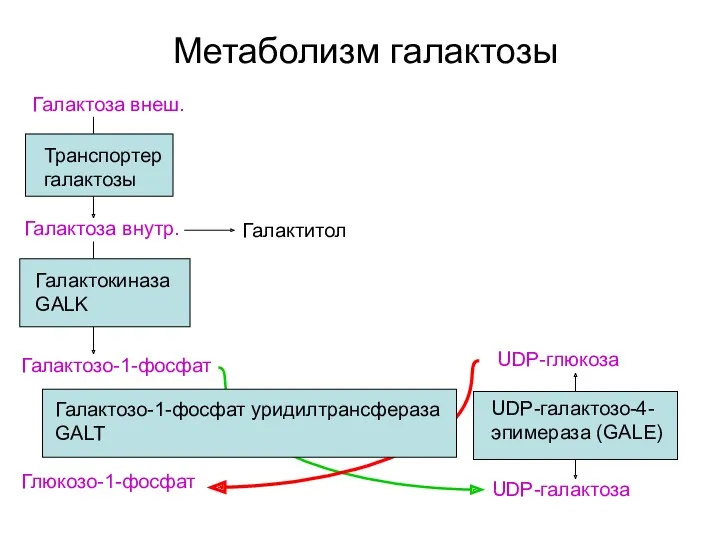
Метаболизм галактозы
Галактоза внеш.
Транспортер галактозы
Галактокиназа GALK
Галактозо-1-фосфат
UDP-галактоза
Глюкозо-1-фосфат
Галактоза внутр.
UDP-глюкоза
UDP-галактозо-4-эпимераза (GALE)
Галактитол
Метаболизм галактозы
Галактоза внеш.
Транспортер галактозы
Галактокиназа GALK
Галактозо-1-фосфат
UDP-галактоза
Глюкозо-1-фосфат
Галактоза внутр.
UDP-глюкоза
UDP-галактозо-4-эпимераза (GALE)
Галактитол
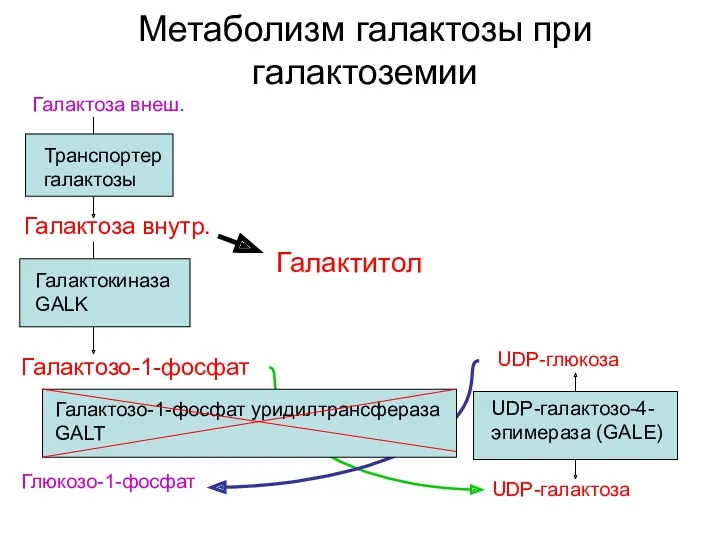
Метаболизм галактозы при галактоземии
Галактоза внеш.
Транспортер галактозы
Галактокиназа GALK
Галактозо-1-фосфат
UDP-галактоза
Глюкозо-1-фосфат
Галактоза внутр.
UDP-глюкоза
UDP-галактозо-4-эпимераза (GALE)
Галактитол
Метаболизм галактозы при галактоземии
Галактоза внеш.
Транспортер галактозы
Галактокиназа GALK
Галактозо-1-фосфат
UDP-галактоза
Глюкозо-1-фосфат
Галактоза внутр.
UDP-глюкоза
UDP-галактозо-4-эпимераза (GALE)
Галактитол
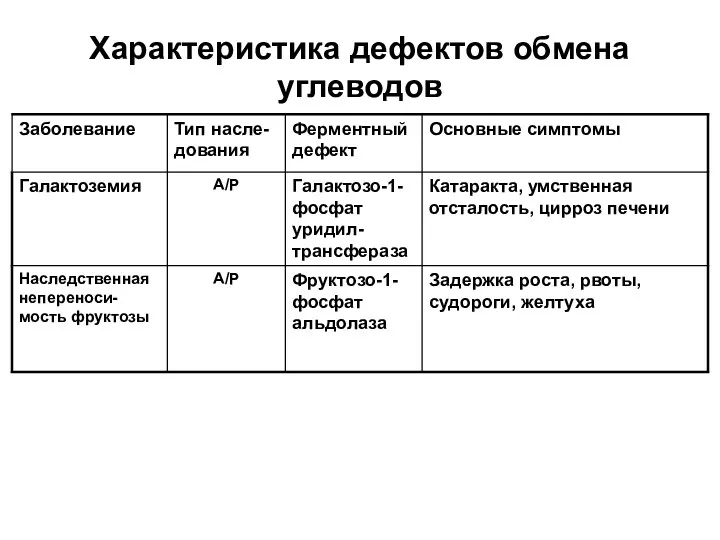
Характеристика дефектов обмена углеводов
Характеристика дефектов обмена углеводов

Нарушения обмена жиров
Нарушения обмена жиров

Множественные ксантомы при семейной гиперхолестеринемии
Множественные ксантомы при семейной гиперхолестеринемии

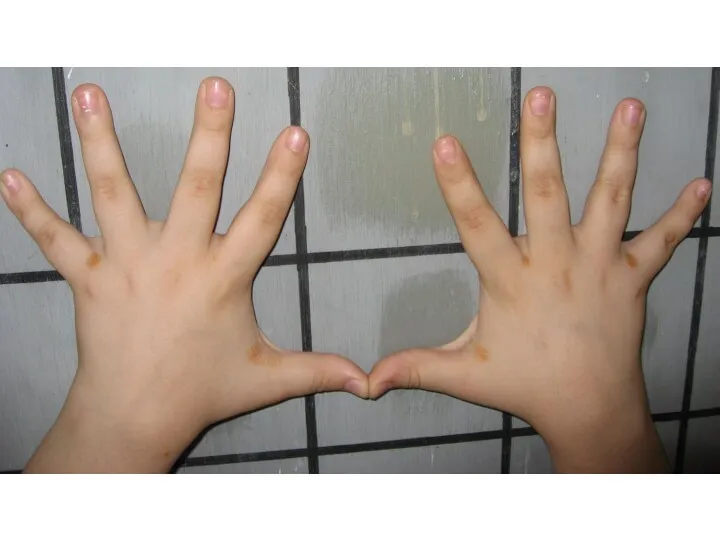

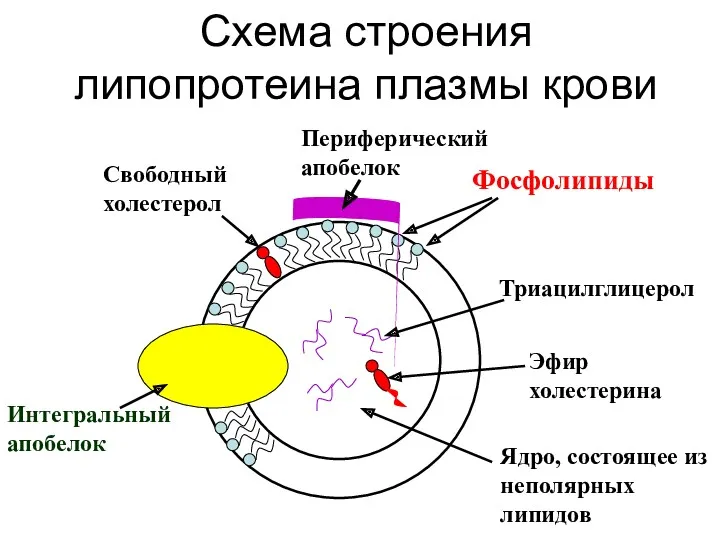
Схема строения липопротеина плазмы крови
Интегральный апобелок
Свободный холестерол
Периферический апобелок
Фосфолипиды
Триацилглицерол
Эфир холестерина
Ядро, состоящее из
Схема строения липопротеина плазмы крови
Интегральный апобелок
Свободный холестерол
Периферический апобелок
Фосфолипиды
Триацилглицерол
Эфир холестерина
Ядро, состоящее из
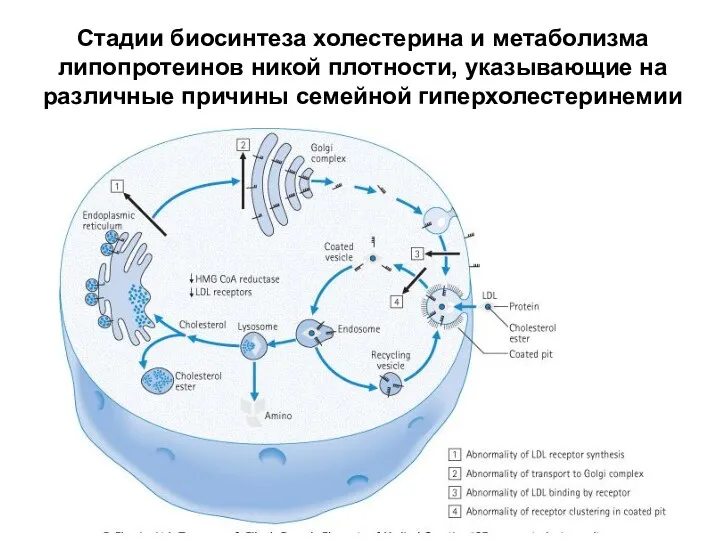
Стадии биосинтеза холестерина и метаболизма липопротеинов никой плотности, указывающие на различные
Стадии биосинтеза холестерина и метаболизма липопротеинов никой плотности, указывающие на различные

Болезни обмена микроэлементов.
Болезни обмена микроэлементов.

Болезнь Вильсона (гепатолентикулярная дегенерация)
Кольцо Кайзера-Флейшера. Отложения меди по краю радужной оболочки
Болезнь Вильсона (гепатолентикулярная дегенерация)
Кольцо Кайзера-Флейшера. Отложения меди по краю радужной оболочки

Скрининг на частые наследственные болезни
Скрининг на частые наследственные болезни

Общие характеристики скрининга
Массовый безотборный характер обследования
Профилактическая направленность
Двухэтапность диагностики
Общие характеристики скрининга
Массовый безотборный характер обследования
Профилактическая направленность
Двухэтапность диагностики

Критерии отбора заболеваний для скрининга
Болезнь без лечения существенно снижает жизнеспособность, ведет
Критерии отбора заболеваний для скрининга
Болезнь без лечения существенно снижает жизнеспособность, ведет

Критерии отбора заболеваний для скрининга
Существуют эффективные методы лечения болезни.
Частота заболевания 1:10
Критерии отбора заболеваний для скрининга
Существуют эффективные методы лечения болезни.
Частота заболевания 1:10

Требования к методам скрининга
Экономичность. Методы диагностики болезни должны быть простыми и
Требования к методам скрининга
Экономичность. Методы диагностики болезни должны быть простыми и

Этапы скрининга
Взятие биологического материала у новорожденных и доставка в лабораторию
Лабораторная просеивающая
Этапы скрининга
Взятие биологического материала у новорожденных и доставка в лабораторию
Лабораторная просеивающая

Нозологические формы, подлежащие скринингу в Российской Федерации
Фенилкетонурия
Галактоземия
Врожденный гипотиреоз
Муковисцидоз
Врожденная гиперплазия коры надпочечников
1:10
Нозологические формы, подлежащие скринингу в Российской Федерации
Фенилкетонурия
Галактоземия
Врожденный гипотиреоз
Муковисцидоз
Врожденная гиперплазия коры надпочечников
1:10

Скрининг на ФКУ
Определение уровня фенилаланина в крови, взятой у новорожденных
Скрининг на ФКУ
Определение уровня фенилаланина в крови, взятой у новорожденных

Скрининг нагалактоземию
Определение уровня галактозы в крови, взятой у новорожденных на 4
Скрининг нагалактоземию
Определение уровня галактозы в крови, взятой у новорожденных на 4

Врожденный гипотиреоз
Гипотиреоз (hypothyreosis; гипо- + анат. glandula thyreoidea щитовидная железа +
Врожденный гипотиреоз
Гипотиреоз (hypothyreosis; гипо- + анат. glandula thyreoidea щитовидная железа +
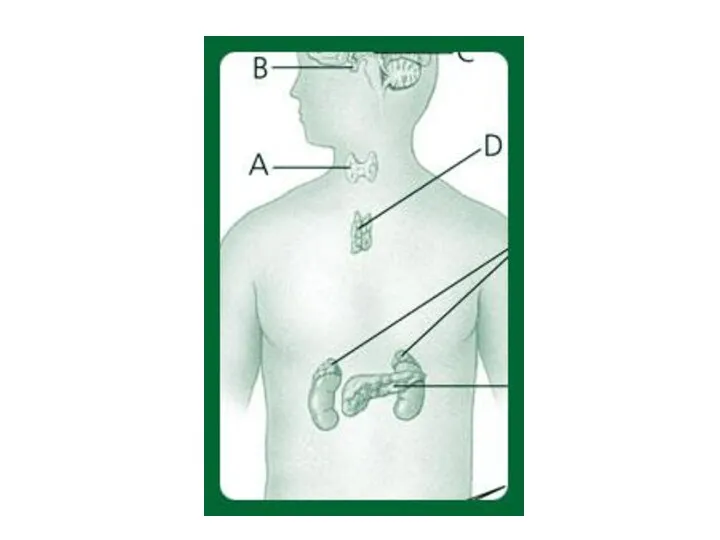

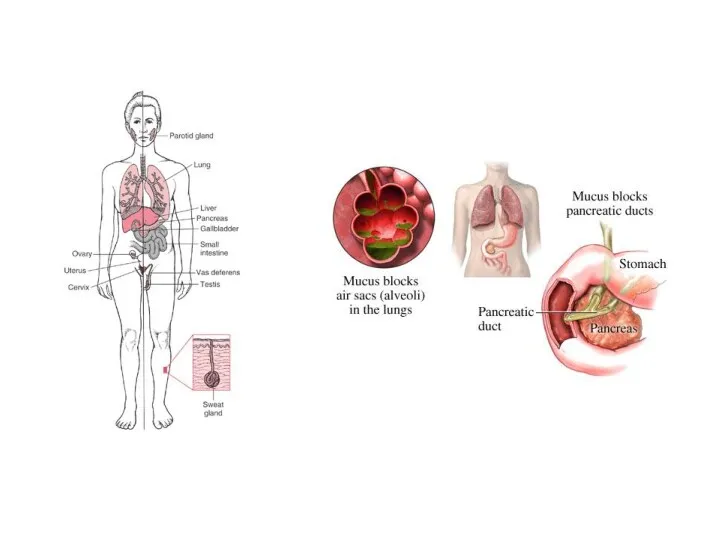
Муковисцидоз
Муковисцидоз (mucoviscidosis; мука- + лат. viscidus липкий + -оз; син.: диспория
Муковисцидоз
Муковисцидоз (mucoviscidosis; мука- + лат. viscidus липкий + -оз; син.: диспория

Адреногенитальный синдром
АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ (врожденная дисфункция коры надпочечников, врожденная гиперплазия коры надпочечников)
Адреногенитальный синдром
АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ (врожденная дисфункция коры надпочечников, врожденная гиперплазия коры надпочечников)
 Полиомиелит. Протекание болезни полиомиелита
Полиомиелит. Протекание болезни полиомиелита Дисфункционалдық жатырдан қан кету
Дисфункционалдық жатырдан қан кету Интерпретация данных методов инструментальной диагностики ЭЭГ психических расстройств, связанных с органическим поражением ЦНС
Интерпретация данных методов инструментальной диагностики ЭЭГ психических расстройств, связанных с органическим поражением ЦНС Временная остановка наружного кровотечения. Ошибки на догоспитальном этапе
Временная остановка наружного кровотечения. Ошибки на догоспитальном этапе Акушерлік перитонит. Жайылған септикалық инфекция
Акушерлік перитонит. Жайылған септикалық инфекция Синдром хронической болезни почек
Синдром хронической болезни почек Балалар ауруларын біріктіріп жүргізу жоспарын құрастыру(жөтел және қиындаған тыныс)
Балалар ауруларын біріктіріп жүргізу жоспарын құрастыру(жөтел және қиындаған тыныс) Иммундық процестердің бұзылуы. Аллергия, анафилаксия, СПИД
Иммундық процестердің бұзылуы. Аллергия, анафилаксия, СПИД Сестринский процесс при гломерулонефритах
Сестринский процесс при гломерулонефритах Гинекологиялық науқастардан анамнез жинау
Гинекологиялық науқастардан анамнез жинау Роль ствола мозга в регуляции двигательных функций
Роль ствола мозга в регуляции двигательных функций Хронический гастрит
Хронический гастрит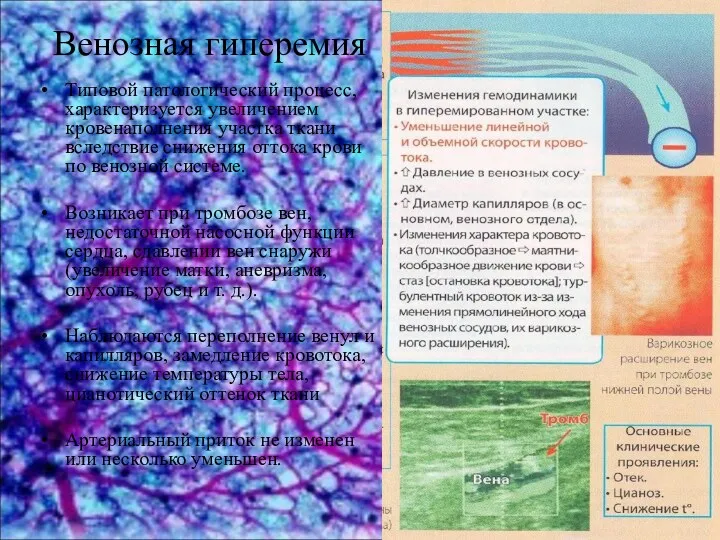 Венозная гиперемия
Венозная гиперемия Электронная медицинская аппаратура
Электронная медицинская аппаратура Лечение заболеваний и травм коленных суставов
Лечение заболеваний и травм коленных суставов Диагностические и профилактические мероприятия болезней вымени
Диагностические и профилактические мероприятия болезней вымени ЛФК при заболеваниях органов пищеварения
ЛФК при заболеваниях органов пищеварения Первая помощь при неотложных состояниях: закон и порядок
Первая помощь при неотложных состояниях: закон и порядок Нарушение кровообращения. Отеки
Нарушение кровообращения. Отеки Лайелл синдромы, Стивен-Джонсон синдромы
Лайелл синдромы, Стивен-Джонсон синдромы Профессиональные нейротоксикозы
Профессиональные нейротоксикозы Азық қорыту жүйесін зерттеу. Азық қабылдау және су ішудің бұзылуы
Азық қорыту жүйесін зерттеу. Азық қабылдау және су ішудің бұзылуы IgA нефропатия. Клиникасы:
IgA нефропатия. Клиникасы: Биохимия почек и мочи. (Лекция 10)
Биохимия почек и мочи. (Лекция 10) Укусы ядовитых животных
Укусы ядовитых животных Көз жасы мүшесінің патологиясы. Дакриоцистит, жас нүктесінің тарылуы, жас нүктесінің сырт айналуы
Көз жасы мүшесінің патологиясы. Дакриоцистит, жас нүктесінің тарылуы, жас нүктесінің сырт айналуы Парвавирусный энтерит собак
Парвавирусный энтерит собак Наследственные болезни обмена веществ
Наследственные болезни обмена веществ