- Наследственные болезни обмена веществ

Содержание
- 2. Наследственные нарушения обмена - большая группу наследственных заболеваний, затрагивающих расстройства метаболизма.
- 3. ЭТИОЛОГИЯ Развитие большинства из них является следствием дефекта единичных генов, кодирующих индивидуальные ферменты, которые обеспечивают превращение
- 4. Как следствие: накопление веществ, обладающих токсическим действием или нарушающих способность синтеза других жизненно важных соединений. Для
- 5. ИСТОРИЯ Термин врожденное нарушение обмена веществ был предложен в начале XX века британским врачом сэром Арчибальдом
- 6. КЛАССИФИКАЦИЯ Нарушения обмена АК Нарушения углеводного обмена Нарушения обмена органических кислот Нарушения окисления жирных кислот и
- 7. НАРУШЕНИЕ ОБМЕНА АК Фенилкетонурия Алкаптонурия Альбинизм Синдром Фанкони Синдром Лоу Гиперлизинемия Орнитинемия Лейциноз
- 8. ФЕНИЛКЕТОНУРИЯ Фенилкетонури́я (фенилпировиноградная олигофрения) — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом
- 9. В большинстве случаев (классическая форма) заболевание связано с резким снижением или полным отсутствием активности печёночного фермента
- 10. Вследствие метаболического блока активируются побочные пути обмена фенилаланина, и в организме происходит накопление его токсичных производных
- 11. ДИАГНОСТИКА Производится полуколичественным тестом или количественным определением фенилаланина в крови. При нелеченных случаях возможно выявление продуктов
- 12. ЛЕЧЕНИЕ При своевременной диагностике патологических изменений можно полностью избежать, если с рождения и до полового созревания
- 14. АЛКАПТОНУРИЯ Алкапто́нури́я — наследственное заболевание, обусловленное выпадением функций оксидазы гомогентизиновой кислоты и характеризующееся расстройством обмена тирозина
- 15. ПАТОГЕНЕЗ Остающаяся в избытке гомогентизиновая кислота превращается алкаптон, который выводится почками. Не полностью экскретируемый мочой алкаптон
- 16. КЛИНИКА выделение у ребенка мочи, быстро темнеющей при стоянии на воздухе, подогревании, подщелачивании. В дальнейшем может
- 17. КЛИНИКА Признаки поражения опорно-двигательного аппарата появляются обычно после 30 лет. Характерно преимущественное поражение крупных суставов нижних
- 18. КЛИИНКА Поражение хрящевой ткани ушных раковин встречается практически у всех больных алкаптонурией в развернутой стадии болезни.
- 20. ДИАГНОСТИКА Наиболее информативным для диагностики алкаптонурии является метод количественного определения гомогентизиновой кислоты и бензохиноуксусной кислоты в
- 21. ЛЕЧЕНИЕ Радикального лечения нет, используется симптоматическая терапия и большие дозы аскорбиновой кислоты.
- 22. АЛЬБИНИЗМ Альбинизм (лат. albus — белый) — врождённое отсутствие пигмента меланина, который придает окраску коже, волосам,
- 23. В настоящее время считается, что причиной альбинизма является отсутствие (или блокада) фермента тирозиназы, необходимой для нормального
- 24. Традиционно альбинизм классифицируют в зависимости от фенотипических проявлений на две большие категории. Глазо-кожный Альбинизм (ГКА) и
- 25. Глазные особенности, общие для всех видов альбинизма, включают в себя: Аномалии рефракции и астигматизм. Нистагм (может
- 27. ЛЕЧЕНИЕ Лечение безуспешно. Восполнить недостаток меланина или предупредить расстройства зрения, связанные с альбинизмом, невозможно. Следует рекомендовать
- 28. СИНДРОМ ФАНКОНИ Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни (перви́чный изоли́рованный синдро́м Фанко́ни, глюко́зо-фосфа́т-ами́новый диабе́т)
- 29. Первые признаки заболевания появляются во втором полугодии жизни — дети вялые, гипотрофичные, аппетит резко снижен, наблюдаются
- 30. Со второго года жизни выявляют отставание физического и ителлектуального развития, происходит генерализованная декальцификация, проявляющаяся костными деформациями
- 31. БИОХИМИЯ снижение уровня кальция в крови; снижение уровня фосфора в крови; повышение уровня щелочной фосфатазы; развитие
- 32. ЛЕЧЕНИЕ Основные принципы — коррекция электролитных нарушений, сдвигов в кислотно-щелочном равновесии, устранение дефицита калия и бикарбонатов.
- 34. НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА Непереносимость лактозы Болезнь Андерсена Болезнь Мак-Ардла Болезнь Гирке. Эссенциальная фруктозурия Галактоземия
- 35. НЕПЕРНОСИМОСТЬ ЛАКТОЗЫ Непереносимость лактозы (или гиполактазия) — термин для описания патологических состояний, вызванных снижением уровня лактазы
- 36. КЛИНИКА избыточный рост и усилением жизнедеятельности микрофлоры кишечника, усваивающей лактозу осмотический эффект непереваренной лактозы в кишечнике
- 37. ДИАГНОСТИКА Пациент принимает 50 граммов лактозы, после чего измеряется содержание водорода в воздухе, выдыхаемом им. Не
- 38. ЛЕЧЕНИЕ Диета Существует возможность принимать фермент лактазу в виде таблеток вместе с молочными продуктами. Таким образом,
- 39. БОЛЕЗНЬ АНДЕРСЕНА Болезнь Андерсена — гликогеноз, семейный цирроз печени, вызванный дефектом фермента амило-(1,4-1,6)-трансглюкозилазы. Заболевание сопровождается избыточным
- 40. КЛИНИКА Первые клинические проявления данной патологии появляются достаточно рано – на первом году жизни ребёнка. Чаще
- 41. КЛИНИКА Одна за другой нарушаются белоксинтетическая, кроветворная, детоксикационная функции печени с развитием соответствующих клинических проявлений. Именно
- 42. ДИАГНОСТИКА Как и при всех гликогенозах, при болезни Андерсена в крови снижается уровень свободной глюкозы, ухудшение
- 43. ЛЕЧЕНИЕ Специфическое лечение при данном гликогенозе не разработано. Прежде всего – борьба с развившимися метаболическими нарушениями,
- 44. БОЛЕЗНЬ МАК-АРДЛА Болезнь Мак-Ардла — гликогеноз, связанный с дефектом мышечной фосфорилазы. Мышцы вынуждены использовать энергию за
- 45. КЛИНИКА Болезнь Мак-Ардля начинается к концу первого десятилетия либо еще позже — вплоть до пятого-шестого десятилетия;
- 46. Вскоре после начала физической нагрузки и появления вышеуказанных симптомов болезни Мак-Ардля состояние улучшается (феномен «второго дыхания»),
- 47. ДИАГНОСТИКА Отсутствие при физической нагрузке увеличения в венозной крови лактата. Диагноз болезни Мак-Ардля может быть подтвержден
- 48. БОЛЕЗНЬ ГИРКЕ Болезнь Гирке — гликогеноз вызванная недостаточностью глюкозо-6-фосфатазы. Недостаточность этого фермента приводит к невозможности превращения
- 49. при болезни Гирке сохраняется способность к преобразованию глюкозы в гликоген и депонированию последнего в тканях различных
- 50. КЛИНИКА 1. неспособность организма поддерживать нормальный уровень глюкозы в крови между приемами пищи; 2. увеличение размеров
- 51. ГИПОГЛИКЕМИЯ Детине спят ночью, даже на втором году жизни. Они могут быть бледными, холодными на ощупь
- 52. ГЕПАТОМЕГАЛИЯ И ПРОБЛЕМЫ С ПЕЧЕНЬЮ Происходит увеличение печени, через накопление гликогена. Кроме печени, гликоген накапливается в
- 54. ЛАКТАТОАЦИДОЗ В результате нарушения глюконеогенеза в организме существенно повышается уровень молочной кислоты (4-10 мМ), даже если
- 55. НАРУШЕНИЕ ФИЗИЧЕСКОГО РАЗВИТИЯ Если болезнь не лечить, то обычным явлением становится задержка процессов физического развития, которая
- 56. РАЗВИТИЕ НЕРВНОЙ СИСТЕМЫ Задержка развития нервной системы является потенциальным вторичным эффектом хронической или рецидивирующей гипогликемии.
- 57. ЛЕЧЕНИЕ Основной целью лечения является предотвращение развития гипогликемии и вторичных метаболических расстройств. Это осуществляется с помощью
- 58. ЛЕЧЕНИЕ Поскольку, при болезни Гирке уровень мочевой кислоты повышается выше 6,5 мг / дл, то для
- 59. НАРУШЕНИЯ ОБМЕНА ПОРФИРИНОВ И БИЛИРУБИНА Синдром Жильбера Синдром Криглера-Найяра Синдром Дубина-Джонсона Нарушение обмена билирубина неуточнённое
- 60. СИНДРОМ ЖИЛЬБЕРА Синдром Жильбера (простая семейная холемия, конституциональная гипербилирубинемия, идиопатическая неконъюгированная гипербилирубинемия, негемолитическая семейная желтуха) —
- 61. Аутосомно-доминантный тип Особенностью является увеличение содержания неконъюгированного билирубина, который не растворим в воде, но хорошо растворим
- 62. МОРФОЛОГИЯ Морфологические изменения в печени характеризуются жировой дистрофией гепатоцитов и накоплением желтовато-коричневого пигмента липофусцина в печёночных
- 63. ЛЕЧЕНИЕ 1.Выведение конъюгированного билирубина (усиленный диурез, активированный уголь как адсорбент билирубина в кишечнике); 2.Связывание уже циркулирующего
- 64. 4.Стремление избежать провоцирующих факторов (инфекции, физические и психические нагрузки, употребление алкоголя и гепатотоксичных лекарств). 5. Противопоказана
- 65. СИНДРОМ ДУБИНА-ДЖОНСОНА Синдром Дабина — Джонсона — энзимопатическая желтуха, редкий пигментный гепатоз, характеризующийся нарушением экскреции связанного
- 66. Синдром Дабина-Джонсона имеет аутосомно-рецессивный тип наследования В крови увеличивается содержание фракции конъюгированного билирубина, в моче —
- 67. МОРФОЛОГИЯ Особенностью этого синдрома является изменение цвета печени: она становится зеленовато-серой или коричневато-чёрной. Макроскопически в ткани
- 69. КЛИНИКА Отмечают повышенную утомляемость плохой аппетит боли в правом подреберье вплоть до колик диарея. Желтуха может
- 70. ДИАГНОСТИКА Лабораторные исследования Обязательные: • общий анализ крови; • общий анализ мочи; • билирубин крови —
- 72. Скачать презентацию

Наследственные нарушения обмена - большая группу наследственных заболеваний, затрагивающих расстройства метаболизма.
Наследственные нарушения обмена - большая группу наследственных заболеваний, затрагивающих расстройства метаболизма.

ЭТИОЛОГИЯ
Развитие большинства из них является следствием дефекта единичных генов, кодирующих индивидуальные
ЭТИОЛОГИЯ
Развитие большинства из них является следствием дефекта единичных генов, кодирующих индивидуальные

Как следствие: накопление веществ, обладающих токсическим действием или нарушающих способность синтеза
Как следствие: накопление веществ, обладающих токсическим действием или нарушающих способность синтеза

ИСТОРИЯ
Термин врожденное нарушение обмена веществ был предложен в начале XX века
ИСТОРИЯ
Термин врожденное нарушение обмена веществ был предложен в начале XX века

КЛАССИФИКАЦИЯ
Нарушения обмена АК
Нарушения углеводного обмена
Нарушения обмена органических кислот
Нарушения окисления жирных кислот
КЛАССИФИКАЦИЯ
Нарушения обмена АК
Нарушения углеводного обмена
Нарушения обмена органических кислот
Нарушения окисления жирных кислот

НАРУШЕНИЕ ОБМЕНА АК
Фенилкетонурия
Алкаптонурия
Альбинизм
Синдром Фанкони
Синдром Лоу
Гиперлизинемия
Орнитинемия
Лейциноз
НАРУШЕНИЕ ОБМЕНА АК
Фенилкетонурия
Алкаптонурия
Альбинизм
Синдром Фанкони
Синдром Лоу
Гиперлизинемия
Орнитинемия
Лейциноз

ФЕНИЛКЕТОНУРИЯ
Фенилкетонури́я (фенилпировиноградная олигофрения) — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма
ФЕНИЛКЕТОНУРИЯ
Фенилкетонури́я (фенилпировиноградная олигофрения) — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма

В большинстве случаев (классическая форма) заболевание связано с резким снижением или
В большинстве случаев (классическая форма) заболевание связано с резким снижением или

Вследствие метаболического блока активируются побочные пути обмена фенилаланина, и в организме
Вследствие метаболического блока активируются побочные пути обмена фенилаланина, и в организме

ДИАГНОСТИКА
Производится полуколичественным тестом или количественным определением фенилаланина в крови.
При нелеченных
ДИАГНОСТИКА
Производится полуколичественным тестом или количественным определением фенилаланина в крови.
При нелеченных

ЛЕЧЕНИЕ
При своевременной диагностике патологических изменений можно полностью избежать, если с рождения
ЛЕЧЕНИЕ
При своевременной диагностике патологических изменений можно полностью избежать, если с рождения

АЛКАПТОНУРИЯ
Алкапто́нури́я — наследственное заболевание, обусловленное выпадением функций оксидазы гомогентизиновой кислоты и характеризующееся
АЛКАПТОНУРИЯ
Алкапто́нури́я — наследственное заболевание, обусловленное выпадением функций оксидазы гомогентизиновой кислоты и характеризующееся

ПАТОГЕНЕЗ
Остающаяся в избытке гомогентизиновая кислота превращается алкаптон, который выводится почками. Не
ПАТОГЕНЕЗ
Остающаяся в избытке гомогентизиновая кислота превращается алкаптон, который выводится почками. Не

КЛИНИКА
выделение у ребенка мочи, быстро темнеющей при стоянии на воздухе, подогревании,
КЛИНИКА
выделение у ребенка мочи, быстро темнеющей при стоянии на воздухе, подогревании,
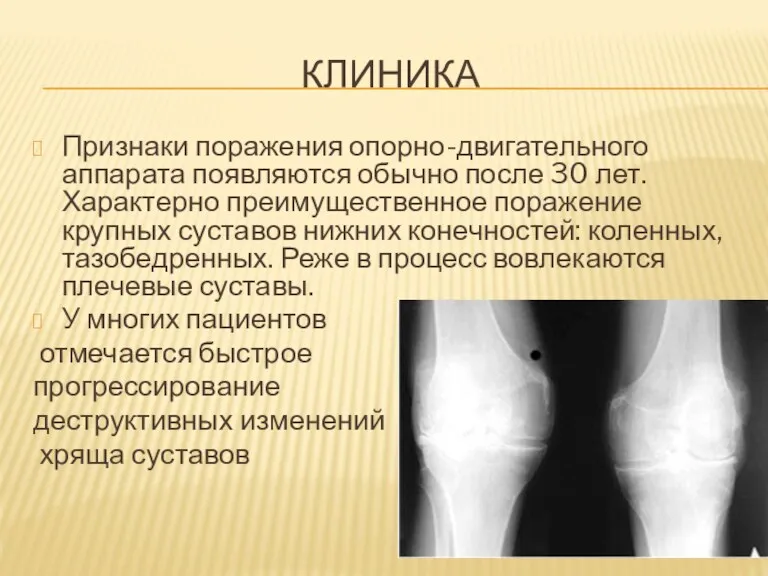
КЛИНИКА
Признаки поражения опорно-двигательного аппарата появляются обычно после 30 лет. Характерно преимущественное
КЛИНИКА
Признаки поражения опорно-двигательного аппарата появляются обычно после 30 лет. Характерно преимущественное

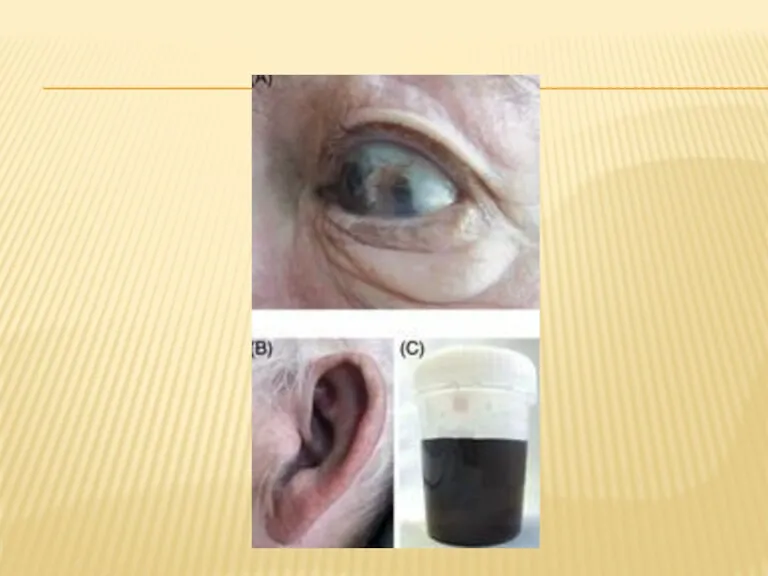
КЛИИНКА
Поражение хрящевой ткани ушных раковин встречается практически у всех больных алкаптонурией
КЛИИНКА
Поражение хрящевой ткани ушных раковин встречается практически у всех больных алкаптонурией

ДИАГНОСТИКА
Наиболее информативным для диагностики алкаптонурии является метод количественного определения гомогентизиновой кислоты
ДИАГНОСТИКА
Наиболее информативным для диагностики алкаптонурии является метод количественного определения гомогентизиновой кислоты

ЛЕЧЕНИЕ
Радикального лечения нет, используется симптоматическая терапия и большие дозы аскорбиновой кислоты.
ЛЕЧЕНИЕ
Радикального лечения нет, используется симптоматическая терапия и большие дозы аскорбиновой кислоты.

АЛЬБИНИЗМ
Альбинизм (лат. albus — белый) — врождённое отсутствие пигмента меланина, который придает окраску
АЛЬБИНИЗМ
Альбинизм (лат. albus — белый) — врождённое отсутствие пигмента меланина, который придает окраску

В настоящее время считается, что причиной альбинизма является отсутствие (или блокада)
В настоящее время считается, что причиной альбинизма является отсутствие (или блокада)

Традиционно альбинизм классифицируют в зависимости от фенотипических проявлений на две большие
Традиционно альбинизм классифицируют в зависимости от фенотипических проявлений на две большие

Глазные особенности, общие для всех видов альбинизма, включают в себя:
Аномалии рефракции
Глазные особенности, общие для всех видов альбинизма, включают в себя:
Аномалии рефракции


ЛЕЧЕНИЕ
Лечение безуспешно. Восполнить недостаток меланина или предупредить расстройства зрения, связанные с
ЛЕЧЕНИЕ
Лечение безуспешно. Восполнить недостаток меланина или предупредить расстройства зрения, связанные с

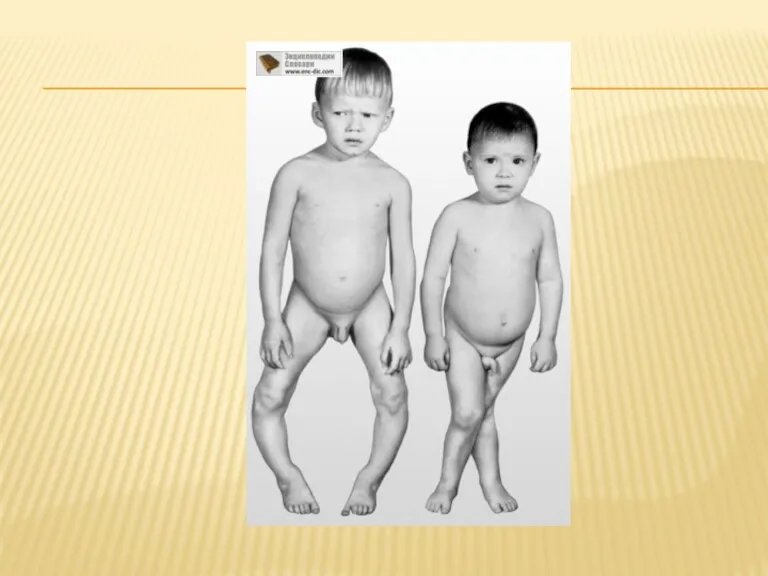
СИНДРОМ ФАНКОНИ
Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни (перви́чный изоли́рованный
СИНДРОМ ФАНКОНИ
Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни (перви́чный изоли́рованный

Первые признаки заболевания появляются во втором полугодии жизни — дети вялые,
Первые признаки заболевания появляются во втором полугодии жизни — дети вялые,

Со второго года жизни выявляют отставание физического и ителлектуального развития, происходит
Со второго года жизни выявляют отставание физического и ителлектуального развития, происходит

БИОХИМИЯ
снижение уровня кальция в крови;
снижение уровня фосфора в крови;
повышение уровня щелочной
БИОХИМИЯ
снижение уровня кальция в крови;
снижение уровня фосфора в крови;
повышение уровня щелочной

ЛЕЧЕНИЕ
Основные принципы — коррекция электролитных нарушений, сдвигов в кислотно-щелочном равновесии, устранение дефицита
ЛЕЧЕНИЕ
Основные принципы — коррекция электролитных нарушений, сдвигов в кислотно-щелочном равновесии, устранение дефицита

НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
Непереносимость лактозы
Болезнь Андерсена
Болезнь Мак-Ардла
Болезнь Гирке.
Эссенциальная фруктозурия
Галактоземия
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА
Непереносимость лактозы
Болезнь Андерсена
Болезнь Мак-Ардла
Болезнь Гирке.
Эссенциальная фруктозурия
Галактоземия

НЕПЕРНОСИМОСТЬ ЛАКТОЗЫ
Непереносимость лактозы (или гиполактазия) — термин для описания патологических состояний, вызванных
НЕПЕРНОСИМОСТЬ ЛАКТОЗЫ
Непереносимость лактозы (или гиполактазия) — термин для описания патологических состояний, вызванных

КЛИНИКА
избыточный рост и усилением жизнедеятельности микрофлоры кишечника, усваивающей лактозу
осмотический эффект
КЛИНИКА
избыточный рост и усилением жизнедеятельности микрофлоры кишечника, усваивающей лактозу
осмотический эффект

ДИАГНОСТИКА
Пациент принимает 50 граммов лактозы, после чего измеряется содержание водорода в
ДИАГНОСТИКА
Пациент принимает 50 граммов лактозы, после чего измеряется содержание водорода в

ЛЕЧЕНИЕ
Диета
Существует возможность принимать фермент лактазу в виде таблеток вместе
ЛЕЧЕНИЕ
Диета
Существует возможность принимать фермент лактазу в виде таблеток вместе

БОЛЕЗНЬ АНДЕРСЕНА
Болезнь Андерсена — гликогеноз, семейный цирроз печени, вызванный дефектом фермента
БОЛЕЗНЬ АНДЕРСЕНА
Болезнь Андерсена — гликогеноз, семейный цирроз печени, вызванный дефектом фермента

КЛИНИКА
Первые клинические проявления данной патологии появляются достаточно рано – на первом
КЛИНИКА
Первые клинические проявления данной патологии появляются достаточно рано – на первом

КЛИНИКА
Одна за другой нарушаются белоксинтетическая, кроветворная, детоксикационная функции печени с развитием
КЛИНИКА
Одна за другой нарушаются белоксинтетическая, кроветворная, детоксикационная функции печени с развитием

ДИАГНОСТИКА
Как и при всех гликогенозах, при болезни Андерсена в крови снижается
ДИАГНОСТИКА
Как и при всех гликогенозах, при болезни Андерсена в крови снижается

ЛЕЧЕНИЕ
Специфическое лечение при данном гликогенозе не разработано. Прежде всего – борьба
ЛЕЧЕНИЕ
Специфическое лечение при данном гликогенозе не разработано. Прежде всего – борьба

БОЛЕЗНЬ МАК-АРДЛА
Болезнь Мак-Ардла — гликогеноз, связанный с дефектом мышечной фосфорилазы.
Мышцы вынуждены
БОЛЕЗНЬ МАК-АРДЛА
Болезнь Мак-Ардла — гликогеноз, связанный с дефектом мышечной фосфорилазы.
Мышцы вынуждены

КЛИНИКА
Болезнь Мак-Ардля начинается к концу первого десятилетия либо еще позже —
КЛИНИКА
Болезнь Мак-Ардля начинается к концу первого десятилетия либо еще позже —

Вскоре после начала физической нагрузки и появления вышеуказанных симптомов болезни Мак-Ардля
Вскоре после начала физической нагрузки и появления вышеуказанных симптомов болезни Мак-Ардля

ДИАГНОСТИКА
Отсутствие при физической нагрузке увеличения в венозной крови лактата.
Диагноз болезни
ДИАГНОСТИКА
Отсутствие при физической нагрузке увеличения в венозной крови лактата.
Диагноз болезни

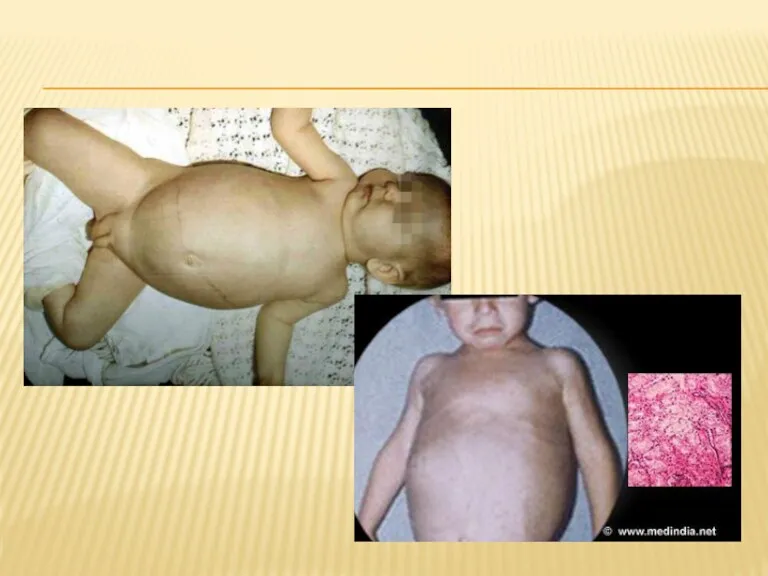
БОЛЕЗНЬ ГИРКЕ
Болезнь Гирке — гликогеноз вызванная недостаточностью глюкозо-6-фосфатазы.
Недостаточность этого фермента приводит к
БОЛЕЗНЬ ГИРКЕ
Болезнь Гирке — гликогеноз вызванная недостаточностью глюкозо-6-фосфатазы.
Недостаточность этого фермента приводит к

при болезни Гирке сохраняется способность к преобразованию глюкозы в гликоген и
при болезни Гирке сохраняется способность к преобразованию глюкозы в гликоген и

КЛИНИКА
1. неспособность организма поддерживать нормальный уровень глюкозы в крови между приемами
КЛИНИКА
1. неспособность организма поддерживать нормальный уровень глюкозы в крови между приемами

ГИПОГЛИКЕМИЯ
Детине спят ночью, даже на втором году жизни. Они могут быть бледными,
ГИПОГЛИКЕМИЯ
Детине спят ночью, даже на втором году жизни. Они могут быть бледными,

ГЕПАТОМЕГАЛИЯ И ПРОБЛЕМЫ С ПЕЧЕНЬЮ
Происходит увеличение печени, через накопление гликогена. Кроме печени,
ГЕПАТОМЕГАЛИЯ И ПРОБЛЕМЫ С ПЕЧЕНЬЮ
Происходит увеличение печени, через накопление гликогена. Кроме печени,

ЛАКТАТОАЦИДОЗ
В результате нарушения глюконеогенеза в организме существенно повышается уровень молочной кислоты
ЛАКТАТОАЦИДОЗ
В результате нарушения глюконеогенеза в организме существенно повышается уровень молочной кислоты

НАРУШЕНИЕ ФИЗИЧЕСКОГО РАЗВИТИЯ
Если болезнь не лечить, то обычным явлением становится
НАРУШЕНИЕ ФИЗИЧЕСКОГО РАЗВИТИЯ
Если болезнь не лечить, то обычным явлением становится

РАЗВИТИЕ НЕРВНОЙ СИСТЕМЫ
Задержка развития нервной системы является потенциальным вторичным эффектом
РАЗВИТИЕ НЕРВНОЙ СИСТЕМЫ
Задержка развития нервной системы является потенциальным вторичным эффектом

ЛЕЧЕНИЕ
Основной целью лечения является предотвращение развития гипогликемии и вторичных метаболических расстройств.
Это
ЛЕЧЕНИЕ
Основной целью лечения является предотвращение развития гипогликемии и вторичных метаболических расстройств.
Это

ЛЕЧЕНИЕ
Поскольку, при болезни Гирке уровень мочевой кислоты повышается
выше 6,5
ЛЕЧЕНИЕ
Поскольку, при болезни Гирке уровень мочевой кислоты повышается
выше 6,5

НАРУШЕНИЯ ОБМЕНА ПОРФИРИНОВ И БИЛИРУБИНА
Синдром Жильбера
Синдром Криглера-Найяра
Синдром Дубина-Джонсона
Нарушение обмена билирубина неуточнённое
НАРУШЕНИЯ ОБМЕНА ПОРФИРИНОВ И БИЛИРУБИНА
Синдром Жильбера
Синдром Криглера-Найяра
Синдром Дубина-Джонсона
Нарушение обмена билирубина неуточнённое

СИНДРОМ ЖИЛЬБЕРА
Синдром Жильбера (простая семейная холемия, конституциональная гипербилирубинемия, идиопатическая неконъюгированная гипербилирубинемия,
СИНДРОМ ЖИЛЬБЕРА
Синдром Жильбера (простая семейная холемия, конституциональная гипербилирубинемия, идиопатическая неконъюгированная гипербилирубинемия,

Аутосомно-доминантный тип
Особенностью является увеличение содержания неконъюгированного билирубина, который не растворим в
Аутосомно-доминантный тип
Особенностью является увеличение содержания неконъюгированного билирубина, который не растворим в

МОРФОЛОГИЯ
Морфологические изменения в печени характеризуются жировой дистрофией гепатоцитов и накоплением желтовато-коричневого
МОРФОЛОГИЯ
Морфологические изменения в печени характеризуются жировой дистрофией гепатоцитов и накоплением желтовато-коричневого

ЛЕЧЕНИЕ
1.Выведение конъюгированного билирубина (усиленный диурез, активированный уголь как адсорбент билирубина в
ЛЕЧЕНИЕ
1.Выведение конъюгированного билирубина (усиленный диурез, активированный уголь как адсорбент билирубина в

4.Стремление избежать провоцирующих факторов (инфекции, физические и психические нагрузки, употребление алкоголя
4.Стремление избежать провоцирующих факторов (инфекции, физические и психические нагрузки, употребление алкоголя

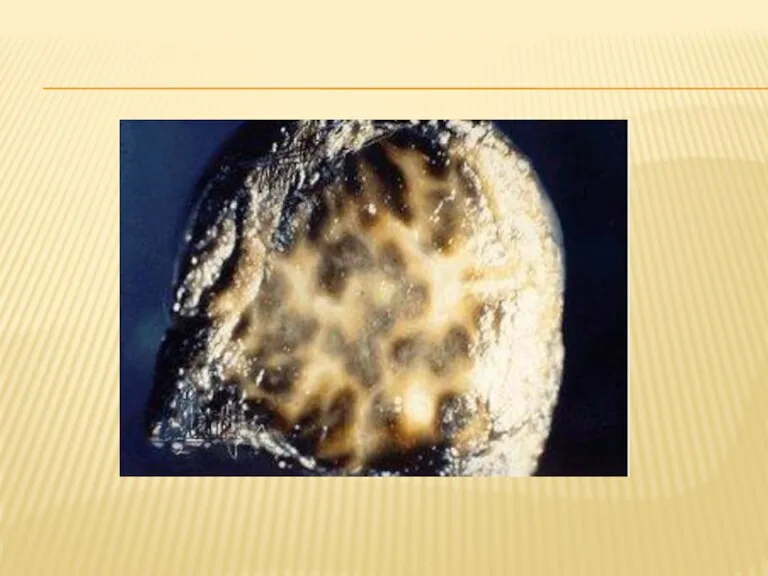
СИНДРОМ ДУБИНА-ДЖОНСОНА
Синдром Дабина — Джонсона — энзимопатическая желтуха, редкий пигментный гепатоз, характеризующийся нарушением
СИНДРОМ ДУБИНА-ДЖОНСОНА
Синдром Дабина — Джонсона — энзимопатическая желтуха, редкий пигментный гепатоз, характеризующийся нарушением

Синдром Дабина-Джонсона имеет аутосомно-рецессивный тип наследования
В крови увеличивается содержание фракции конъюгированного
Синдром Дабина-Джонсона имеет аутосомно-рецессивный тип наследования
В крови увеличивается содержание фракции конъюгированного

МОРФОЛОГИЯ
Особенностью этого синдрома является изменение цвета печени: она становится зеленовато-серой или
МОРФОЛОГИЯ
Особенностью этого синдрома является изменение цвета печени: она становится зеленовато-серой или

КЛИНИКА
Отмечают повышенную утомляемость
плохой аппетит
боли в правом подреберье вплоть до колик
КЛИНИКА
Отмечают повышенную утомляемость
плохой аппетит
боли в правом подреберье вплоть до колик

ДИАГНОСТИКА
Лабораторные исследования
Обязательные:
• общий анализ крови;
• общий анализ мочи;
• билирубин крови — повышение
ДИАГНОСТИКА
Лабораторные исследования
Обязательные:
• общий анализ крови;
• общий анализ мочи;
• билирубин крови — повышение
 Эндокринологическая аллея. Остановка Музей изобразительных искусств
Эндокринологическая аллея. Остановка Музей изобразительных искусств Cтандарты в производстве медицинских изделий
Cтандарты в производстве медицинских изделий Язвенная болезнь желудка и 12-перстной кишки
Язвенная болезнь желудка и 12-перстной кишки Поражение мозжечка
Поражение мозжечка Фагоцитоз. Микрофаги и макрофаги
Фагоцитоз. Микрофаги и макрофаги Аяқ іріңі жаралары бар науқастарды емдеуде, озонды оттегі қоспаларын пайдалану тиімділігін бағалау
Аяқ іріңі жаралары бар науқастарды емдеуде, озонды оттегі қоспаларын пайдалану тиімділігін бағалау Информация для младшего мед. персонала противотуберкулёзного стационара
Информация для младшего мед. персонала противотуберкулёзного стационара Гипогликемия и гипергликемия у новорожденных
Гипогликемия и гипергликемия у новорожденных Гигиенические требования к выбору и планировке больничного участка. Системы строительства больниц, их преимущества и недостатки
Гигиенические требования к выбору и планировке больничного участка. Системы строительства больниц, их преимущества и недостатки Легионеллы. Морфология
Легионеллы. Морфология Стоматология. Жалобы на эстетический дефект
Стоматология. Жалобы на эстетический дефект Патологиялық анатомия. Ісіктер жөнінде жалпы ілім
Патологиялық анатомия. Ісіктер жөнінде жалпы ілім Местная хирургическая патология и ее лечение (раны)
Местная хирургическая патология и ее лечение (раны) Поздний гестоз беременных
Поздний гестоз беременных Врожденный сифилис
Врожденный сифилис Caries
Caries Туберкулинодиагностика
Туберкулинодиагностика Технология гомеопатических таблеток
Технология гомеопатических таблеток Оказание медицинской помощи при одноплодных родах в затылочном предлежании во внебольничных условиях
Оказание медицинской помощи при одноплодных родах в затылочном предлежании во внебольничных условиях Проблема биосовместимости (лекция 3)
Проблема биосовместимости (лекция 3) Суппозитории
Суппозитории Өкпе туберкулезінің рентгенодиагностикасы
Өкпе туберкулезінің рентгенодиагностикасы Гепатит В
Гепатит В Научно – обоснованная медицинская практика. Поиск доказательной информации. Базы данных
Научно – обоснованная медицинская практика. Поиск доказательной информации. Базы данных Гематологиялық анализаторлар. Анализ нәтижелеріне талдау жасау
Гематологиялық анализаторлар. Анализ нәтижелеріне талдау жасау Ротавирусы. Энтеровирусы. Рабдовирусы. Тогавирусы
Ротавирусы. Энтеровирусы. Рабдовирусы. Тогавирусы Орталық жүйке жүйесінің ноцецептивтік жүйесі. Неврологиядағы ауырсыну синдромы
Орталық жүйке жүйесінің ноцецептивтік жүйесі. Неврологиядағы ауырсыну синдромы Владимир Петрович Филатов 1875 – 1956
Владимир Петрович Филатов 1875 – 1956