- Congenital Adrenal Hyperplasia

Содержание
- 2. What is CAH? It is a familial disorder of adrenal steroid biosynthesis due to adrenal enzyme
- 3. Adrenal steroids biosynthesis
- 4. 21-Hydroxylase deficiency The most common CAH type reduced production of cortisol and aldosterone and increased production
- 5. Classical 21-hydroxylase deficiency Salt-wasting form (75%): unable to synthesize adequate amounts of cortisol and aldosterone, lose
- 6. Non-classical 21-hydroxylase deficiency Usually is mild and manifest as an androgen excess later in life. Aldosterone
- 7. Diagnosis of 21-hydroxylase deficiency Screening: high blood level of 17-OH Progesterone 250 MKG SYNACTHEN TEST: high
- 8. 11-hydroxylase (CYP11B1) deficiency 11-hydroxylase Hypertension and hypokalemia occur because of accumulation of 11-deoxycorticosterone, a potent mineralocorticoid,
- 9. 17-hydroxylase (CYP11B1) deficiency 17α-hydroxylase Hypertension and hypokalemia occur due to accumulation of aldosterone No female virilizaton
- 10. Diagnosis 11-hydroxylase deficiency 250 MKG SYNACTHEN TEST: high serum level of 11-deoxycorticosterone and 11-deoxycortisol elevated 24-hour
- 11. TREATMENT PRINCIPLES In classical CAH treatment is life-long Treatment goals are: to maintain growth velocity &
- 12. Prenatal diagnosis and treatment Done by chorionic villus sampling at 8-12 wk & amniocentesis at 18-20
- 14. Скачать презентацию

What is CAH?
It is a familial disorder of adrenal steroid biosynthesis
What is CAH?
It is a familial disorder of adrenal steroid biosynthesis
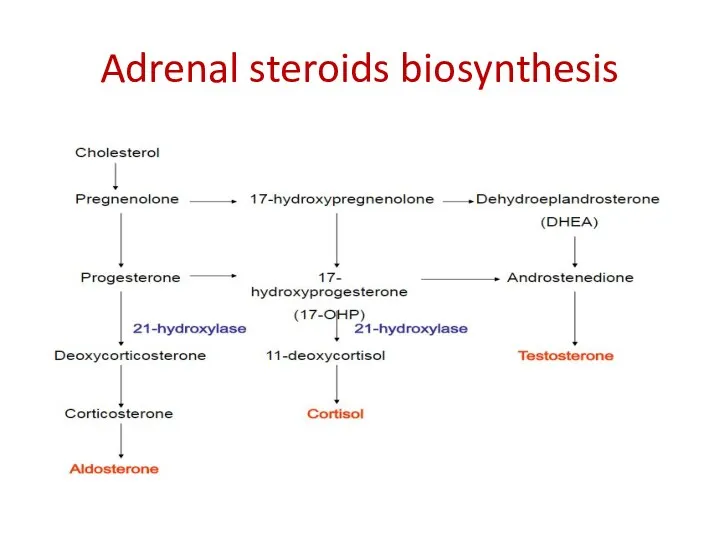
Adrenal steroids biosynthesis
Adrenal steroids biosynthesis
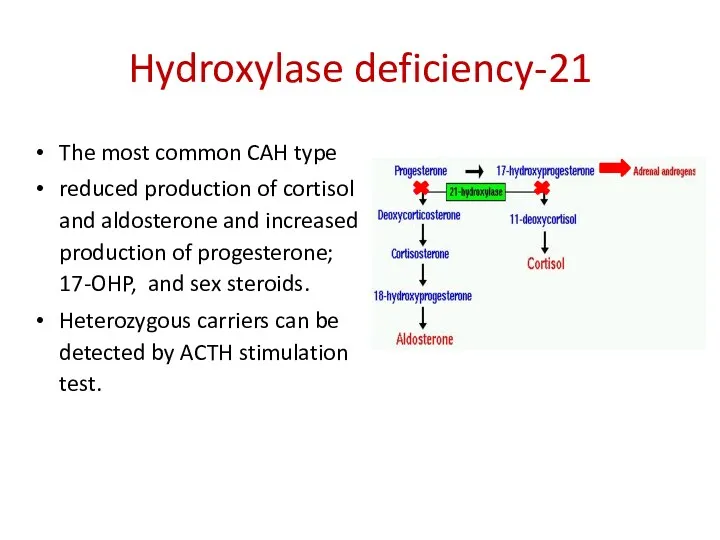
21-Hydroxylase deficiency
The most common CAH type
reduced production of cortisol and
21-Hydroxylase deficiency
The most common CAH type
reduced production of cortisol and
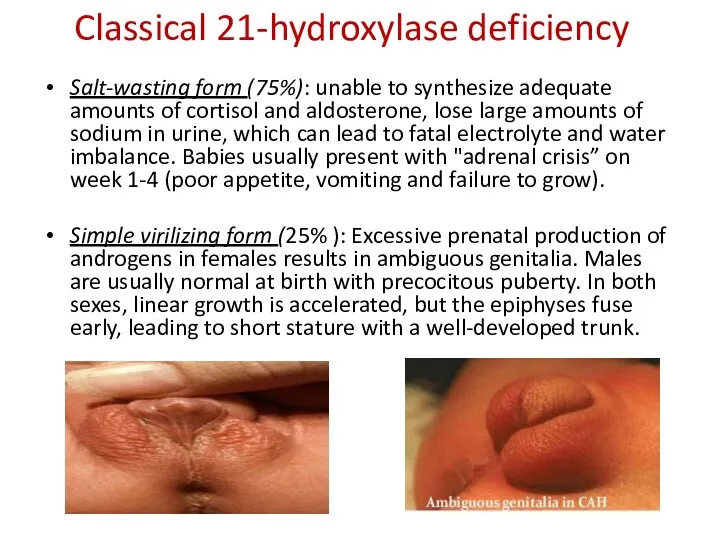
Classical 21-hydroxylase deficiency
Salt-wasting form (75%): unable to synthesize adequate amounts of
Classical 21-hydroxylase deficiency
Salt-wasting form (75%): unable to synthesize adequate amounts of

Non-classical 21-hydroxylase deficiency
Usually is mild and manifest as an androgen excess
Non-classical 21-hydroxylase deficiency
Usually is mild and manifest as an androgen excess

Diagnosis of 21-hydroxylase deficiency
Screening: high blood level of 17-OH Progesterone
250 MKG
Diagnosis of 21-hydroxylase deficiency
Screening: high blood level of 17-OH Progesterone
250 MKG
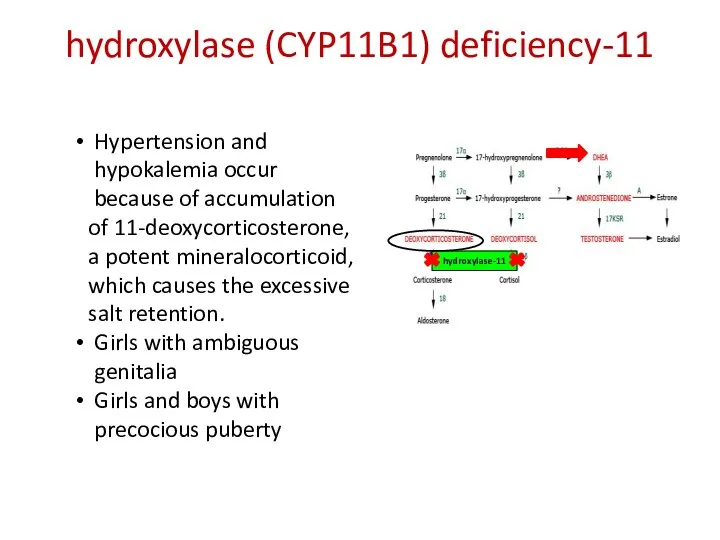
11-hydroxylase (CYP11B1) deficiency
11-hydroxylase
Hypertension and hypokalemia occur
because of accumulation
11-hydroxylase (CYP11B1) deficiency
11-hydroxylase
Hypertension and hypokalemia occur
because of accumulation
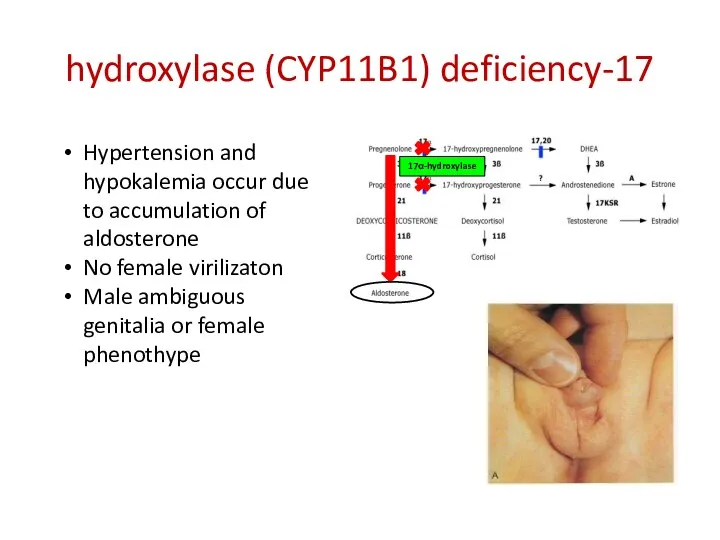
17-hydroxylase (CYP11B1) deficiency
17α-hydroxylase
Hypertension and hypokalemia occur due to accumulation of aldosterone
No
17-hydroxylase (CYP11B1) deficiency
17α-hydroxylase
Hypertension and hypokalemia occur due to accumulation of aldosterone
No

Diagnosis
11-hydroxylase deficiency
250 MKG SYNACTHEN TEST: high serum level of
Diagnosis
11-hydroxylase deficiency
250 MKG SYNACTHEN TEST: high serum level of

TREATMENT PRINCIPLES
In classical CAH treatment is life-long
Treatment goals are:
to maintain
TREATMENT PRINCIPLES
In classical CAH treatment is life-long
Treatment goals are:
to maintain

Prenatal diagnosis and treatment
Done by chorionic villus sampling at 8-12 wk
Prenatal diagnosis and treatment
Done by chorionic villus sampling at 8-12 wk
 Ортопедиялық (ортодентия tel tedavi̇si̇) стоматологияда қолданылатын полимерлер
Ортопедиялық (ортодентия tel tedavi̇si̇) стоматологияда қолданылатын полимерлер Сүйектің эктопиялық дамуы
Сүйектің эктопиялық дамуы Гендік терапия
Гендік терапия Гестационный сахарный диабет
Гестационный сахарный диабет Средства, влияющие на функции органов дыхания
Средства, влияющие на функции органов дыхания Основы онкологии
Основы онкологии Этика и медицинская деонтология. Биоэтика
Этика и медицинская деонтология. Биоэтика Первая помощь при кровотечении
Первая помощь при кровотечении Организация санитарно-противоэпидемического обеспечения и медицинского снабжения в чрезвычайных ситуациях
Организация санитарно-противоэпидемического обеспечения и медицинского снабжения в чрезвычайных ситуациях Медицинская информатика: основы защиты данных. Лекция 3
Медицинская информатика: основы защиты данных. Лекция 3 Қант диабетінің емі
Қант диабетінің емі 1 жасқа дейінгі балаларды табиғи тамақтандыру
1 жасқа дейінгі балаларды табиғи тамақтандыру Туберкулинді диагностика
Туберкулинді диагностика Экопаталогии. Экология человека
Экопаталогии. Экология человека Основные понятия и категории социальной медицины
Основные понятия и категории социальной медицины Коррекция кальций-фосфорного обмена у пациентов на гемодиализе
Коррекция кальций-фосфорного обмена у пациентов на гемодиализе Остеоартроз (остеоартрит)
Остеоартроз (остеоартрит) Опыт хирургического лечения опухолей надпочечников у детей
Опыт хирургического лечения опухолей надпочечников у детей Микобактерии – возбудители микобактериозов, туберкулеза и лепры
Микобактерии – возбудители микобактериозов, туберкулеза и лепры Өңештің бөгде заттары
Өңештің бөгде заттары Клінічні та ЕКГ-ознаки порушень збудливості та провідності
Клінічні та ЕКГ-ознаки порушень збудливості та провідності Преждевременная отслойка нормально расположенной плаценты
Преждевременная отслойка нормально расположенной плаценты Общий уход за больными в предоперационный и послеоперационный период
Общий уход за больными в предоперационный и послеоперационный период Ми қыртысы. Ми қыртысының цитоархитектоникасы
Ми қыртысы. Ми қыртысының цитоархитектоникасы Психология сложного дефекта
Психология сложного дефекта Расслоение аорты, диагностика, неотложная помощь
Расслоение аорты, диагностика, неотложная помощь Социофобия. Панические атаки. Агрофобия. ВСД
Социофобия. Панические атаки. Агрофобия. ВСД Пороки митрального клапана
Пороки митрального клапана