- Фенилкетонурия. Нарушения обмена триптофана

Содержание
- 2. Фенилкетонурия – наиболее распространенное нарушение обмена аминокислот. В среднем фенилкетонурии подвержен 1 из 8000 человек. В
- 3. Фенилкетонурия проявляется на первом году жизни. Основными симптомами в этом возрасте являются: вялость ребенка; отсутствие интереса
- 4. Диагностика скриниг-метод (новорожденным детям в роддоме проводят экспресс-тесты на содержание фенилаланина) дополнительные исследования крови и моче
- 5. Лечение фенилкетонурии Самым эффективным и распространенным способом лечения, является элиминационная диета: диета с исключением продуктов, содержащих
- 6. Назначаются комплексы из витаминов и минералов При судорожных припадках, необходимо применение антиконвульсантов Показан массаж, лечебная физкультура
- 7. Новые направления в лечении фенилкетонурии использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до
- 8. Нарушения обмена триптофана Триптофан – незаменимая аминокислота. Образующийся при расщеплении белков триптофан через кишечную стенку всасывается
- 9. Болезнь Гартнепа Индиканурия генетическое изменение транспортной функции клеток слизистой оболочки кишечника и проксимальных отделов почечных канальцев.
- 11. Скачать презентацию

Фенилкетонурия – наиболее распространенное нарушение обмена аминокислот. В среднем фенилкетонурии подвержен
Фенилкетонурия – наиболее распространенное нарушение обмена аминокислот. В среднем фенилкетонурии подвержен

Фенилкетонурия проявляется на первом году жизни.
Основными симптомами в этом возрасте
Фенилкетонурия проявляется на первом году жизни.
Основными симптомами в этом возрасте

Диагностика
скриниг-метод (новорожденным детям в роддоме проводят экспресс-тесты на содержание фенилаланина)
дополнительные исследования
Диагностика
скриниг-метод (новорожденным детям в роддоме проводят экспресс-тесты на содержание фенилаланина)
дополнительные исследования

Лечение фенилкетонурии
Самым эффективным и распространенным способом лечения, является элиминационная диета: диета
Лечение фенилкетонурии
Самым эффективным и распространенным способом лечения, является элиминационная диета: диета

Назначаются комплексы из витаминов и минералов
При судорожных припадках, необходимо применение антиконвульсантов
Показан
Назначаются комплексы из витаминов и минералов
При судорожных припадках, необходимо применение антиконвульсантов
Показан

Новые направления в лечении фенилкетонурии
использование заместительной терапии фенилаланинлиазой (PAL) – растительным
Новые направления в лечении фенилкетонурии
использование заместительной терапии фенилаланинлиазой (PAL) – растительным

Нарушения обмена триптофана
Триптофан – незаменимая аминокислота. Образующийся при расщеплении белков триптофан
Нарушения обмена триптофана
Триптофан – незаменимая аминокислота. Образующийся при расщеплении белков триптофан
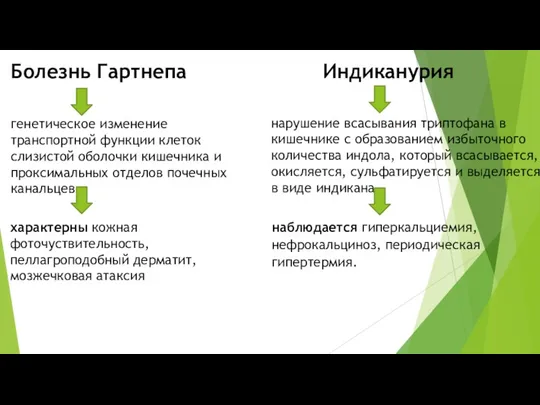
Болезнь Гартнепа
Индиканурия
генетическое изменение транспортной функции клеток слизистой оболочки кишечника и проксимальных
Болезнь Гартнепа
Индиканурия
генетическое изменение транспортной функции клеток слизистой оболочки кишечника и проксимальных
 Неотложная помощь при химических ожогах
Неотложная помощь при химических ожогах Фарфор ( стоматологическая керамика) и диоксид циркония
Фарфор ( стоматологическая керамика) и диоксид циркония Эпилепсия. Классификация припадков. Первая помощь
Эпилепсия. Классификация припадков. Первая помощь Кора головного мозку
Кора головного мозку Тығыз талшықты дәнекер тіні
Тығыз талшықты дәнекер тіні Нейропатия лицевого нерва
Нейропатия лицевого нерва Организационные основы внедрения профилактики стоматологических заболеваний у детей. (Лекция 6)
Организационные основы внедрения профилактики стоматологических заболеваний у детей. (Лекция 6) Сахарный диабет 1 и 2 типа
Сахарный диабет 1 и 2 типа Оценка тяжести пациента
Оценка тяжести пациента Грыжи передней брюшной стенки
Грыжи передней брюшной стенки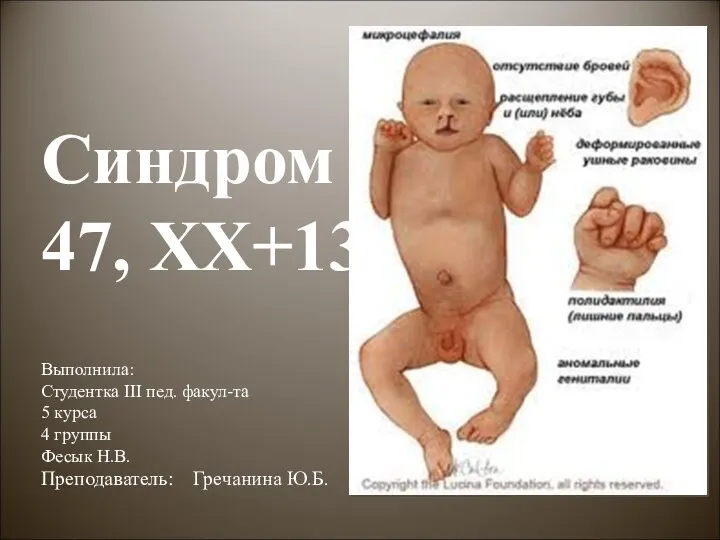 Синдром Патау
Синдром Патау Қант диабеті этиология патогенезі
Қант диабеті этиология патогенезі Первая помощь при ожогах
Первая помощь при ожогах Андрологія. Ветеринарна андрологія
Андрологія. Ветеринарна андрологія Система реабилитации детей с врожденными расщелинами губы и неба
Система реабилитации детей с врожденными расщелинами губы и неба Аралас Т- және В-иммундық тапшылықтар
Аралас Т- және В-иммундық тапшылықтар Дифференциальный диагноз анемий
Дифференциальный диагноз анемий Анатомия головы и шеи
Анатомия головы и шеи Огнестрельные и закрытые повреждения конечностей и суставов
Огнестрельные и закрытые повреждения конечностей и суставов Хроническая свинцовая интоксикация (сатурнизм)
Хроническая свинцовая интоксикация (сатурнизм) ИВЛ-основы
ИВЛ-основы Сердечно-сосудистая хирургия. Сосудистые швы. Аорторафия
Сердечно-сосудистая хирургия. Сосудистые швы. Аорторафия Моногенді аурулар
Моногенді аурулар Невропатология как наука
Невропатология как наука Кариес или зачем лечить молочные зубы
Кариес или зачем лечить молочные зубы Физиология почек
Физиология почек Высшая нервная деятельность
Высшая нервная деятельность Дәлелді медицина мамандарының қоғамы туралы түсінік. ТМД және біздің елдегі дәлелді медицина орталықтары
Дәлелді медицина мамандарының қоғамы туралы түсінік. ТМД және біздің елдегі дәлелді медицина орталықтары