- Генетические дефекты и синдромальные заболевания в гинекологии

Содержание
- 2. Пороки развития половых органов составляют 14% от всех врожденных аномалий с увеличением в 10 раз у
- 3. Класс I - атрезия девственной плевы (варианты строения девственной плевы). Класс II - полная или неполная
- 4. Аплазия влагалища и матки (синдром Майера-Рокитанского-Кюстера-Хауйзера - СМРКХ) Является крайней степенью врожденного дисморфогенеза среди всех случаев
- 6. СМРКХ характеризуется физиологически развитыми вторичными половыми признаками (женский фенотип), нормальным женским кариотипом (46, XX), врожденным отсутствием
- 7. Патогенез Ген НОХА10 ответственен за развитие матки НОХА 11 - нижней сегмент матки и шейку матки
- 10. Синдром Шерешевского-Тёрнера (СШТ) — это хромосомное нарушение, связанное с полной или частичной моносомией по X хромосоме,
- 11. Патогенез Синдром Шерешевского-Тёрнера на цитогенетическом уровне характеризуется моносомией по Х хромосоме, присутствием аномальной Х хромосомы или
- 13. Клиника 1. Низкоролость 2. Крыловидные складки на шее 3. Гипогонадизм 4. Лимфедема 5. Патология сердечно-сосудистой системы
- 14. Диагностика Обычно СШТ диагностируется у девочек в раннем детстве, когда обнаруживается задержка роста и прочие симптомы,
- 15. Лечение СШТ полностью не излечивается. Известные методы лечения являются симптоматическими. Применяется соматотропная гормональная терапия для коррекции
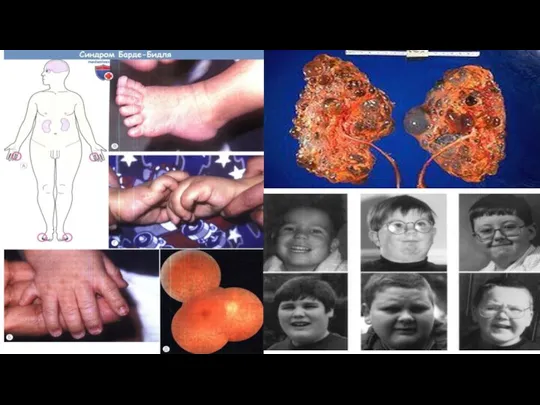
- 16. Синдром Барде-Бидля (Лоуренса- Муна) Аутосомно-рецесивное заболевание, проявляющееся пигментной дегенерацией сетчатки, ожирением, полидактилией, гипогенитализмом и умственной отсталостью.
- 17. Симптомы Из основных симптомов наиболее часто встречаются: пигментный ретинит и другие изменения сетчатки (93%) ожирение (90%)
- 18. Патогенез Синдром Барде-Бидля принято считать аутосомно-рецесивным заболеванием, однако доказано, что для клинической манифестации некоторых форм СББ,
- 20. Синдром Мак-Кьюна— Олбрайта— Брайцева (синдром МОБ, McCune–Albright–Braitsev syndrome) или синдром Олбрайта— Мак-Кьюна— Стернберга (Albright–McCune–Sternberg syndrome), как
- 21. Клинические проявления синдрома: – асимметричные гиперпигментированные пятна цвета кофе с молоком (лентиго), обычно на коже груди,
- 22. Патогенез Синдром МОБ обусловлен соматической мутацией гена GNASI, расположенного на длинном плече 20-й хромосомы, на ранних
- 23. Течение и прогноз Заболевание клинически неоднородное. Наряду со случаями классической триады признаков, существуют атипичные и неполные
- 25. Скачать презентацию

Пороки развития половых органов составляют 14% от всех врожденных аномалий
Пороки развития половых органов составляют 14% от всех врожденных аномалий

Класс I - атрезия девственной плевы (варианты строения девственной плевы).
Класс
Класс I - атрезия девственной плевы (варианты строения девственной плевы).
Класс

Аплазия влагалища и матки (синдром Майера-Рокитанского-Кюстера-Хауйзера - СМРКХ)
Является крайней степенью врожденного
Аплазия влагалища и матки (синдром Майера-Рокитанского-Кюстера-Хауйзера - СМРКХ)
Является крайней степенью врожденного
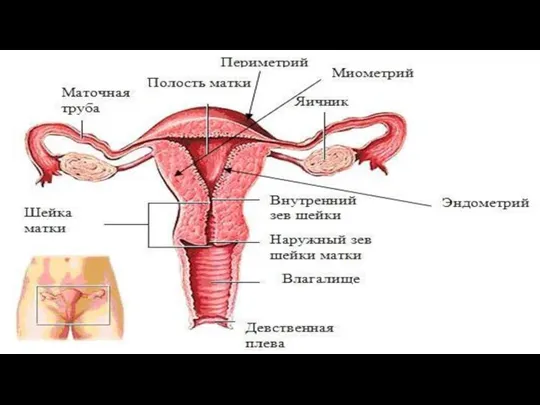
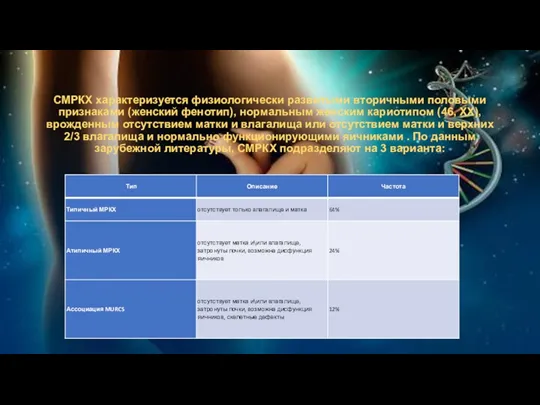
СМРКХ характеризуется физиологически развитыми вторичными половыми признаками (женский фенотип), нормальным женским
СМРКХ характеризуется физиологически развитыми вторичными половыми признаками (женский фенотип), нормальным женским
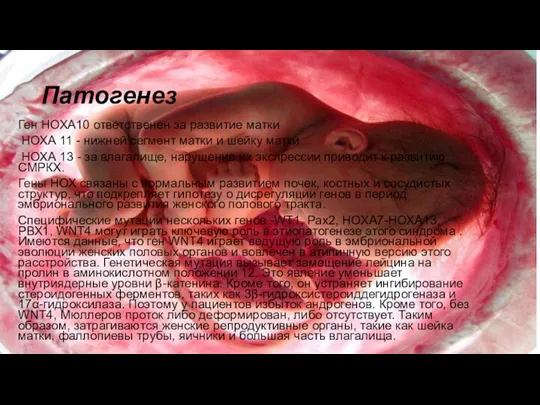
Патогенез
Ген НОХА10 ответственен за развитие матки
НОХА 11 - нижней сегмент
Патогенез
Ген НОХА10 ответственен за развитие матки
НОХА 11 - нижней сегмент
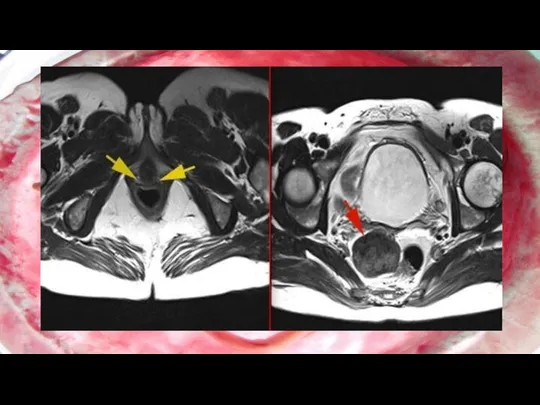
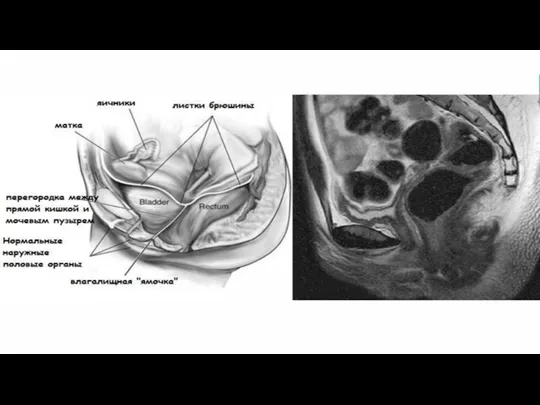
Синдром Шерешевского-Тёрнера (СШТ) — это хромосомное нарушение, связанное с полной или
Синдром Шерешевского-Тёрнера (СШТ) — это хромосомное нарушение, связанное с полной или

Патогенез
Синдром Шерешевского-Тёрнера на цитогенетическом уровне характеризуется моносомией по Х хромосоме,
Патогенез Синдром Шерешевского-Тёрнера на цитогенетическом уровне характеризуется моносомией по Х хромосоме,

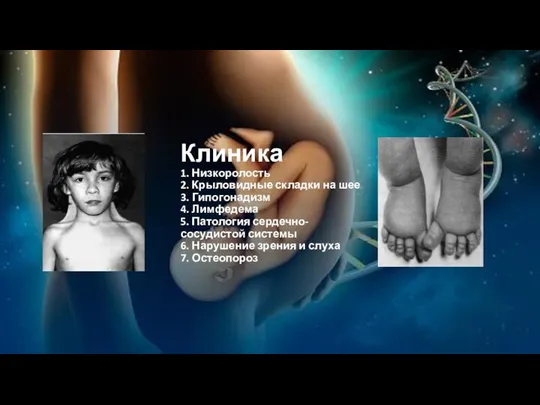
Клиника
1. Низкоролость
2. Крыловидные складки на шее
3. Гипогонадизм
4. Лимфедема
5. Патология сердечно-сосудистой системы
6.
Клиника 1. Низкоролость 2. Крыловидные складки на шее 3. Гипогонадизм 4. Лимфедема 5. Патология сердечно-сосудистой системы 6.

Диагностика
Обычно СШТ диагностируется у девочек в раннем детстве, когда обнаруживается задержка
Диагностика
Обычно СШТ диагностируется у девочек в раннем детстве, когда обнаруживается задержка

Лечение
СШТ полностью не излечивается. Известные методы лечения являются симптоматическими.
Применяется соматотропная гормональная
Лечение
СШТ полностью не излечивается. Известные методы лечения являются симптоматическими.
Применяется соматотропная гормональная

Синдром Барде-Бидля (Лоуренса- Муна)
Аутосомно-рецесивное заболевание, проявляющееся пигментной дегенерацией сетчатки, ожирением, полидактилией, гипогенитализмом и умственной отсталостью. Синдром описан в 1866 г. Лоренсом и Муном как сочетание пигментной дистрофии сетчатки с гипогенитализмом, ожирением и умственной отсталостью, а в 1920 г. Барде и Бидлем, но с добавлением к указанному симптомокомплексу полидактилии. Зафиксировано немногим более 500 больных. Популяционная частота в Европе среди новорожденных невысокая 1: 160 000.
Характеризуется большой вариабельностью проявлений. Чаще наблюдается 3 или 4 признака (неполная форма), реже — пять (полная форма).
Синдром Барде-Бидля (Лоуренса- Муна)
Аутосомно-рецесивное заболевание, проявляющееся пигментной дегенерацией сетчатки, ожирением, полидактилией, гипогенитализмом и умственной отсталостью. Синдром описан в 1866 г. Лоренсом и Муном как сочетание пигментной дистрофии сетчатки с гипогенитализмом, ожирением и умственной отсталостью, а в 1920 г. Барде и Бидлем, но с добавлением к указанному симптомокомплексу полидактилии. Зафиксировано немногим более 500 больных. Популяционная частота в Европе среди новорожденных невысокая 1: 160 000.
Характеризуется большой вариабельностью проявлений. Чаще наблюдается 3 или 4 признака (неполная форма), реже — пять (полная форма).
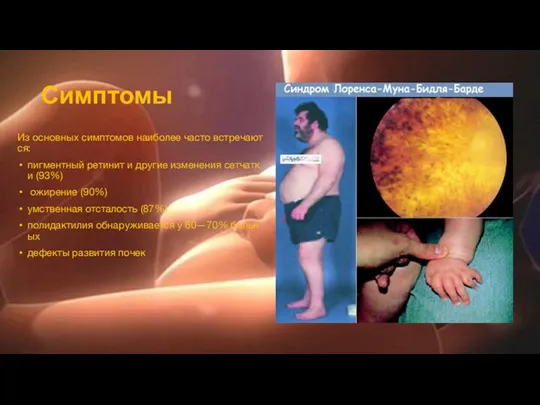
Симптомы
Из основных симптомов наиболее часто встречаются:
пигментный ретинит и другие изменения сетчатки (93%)
ожирение (90%)
умственная отсталость (87%)
полидактилия обнаруживается у 60—70% больных
дефекты развития почек
Симптомы
Из основных симптомов наиболее часто встречаются:
пигментный ретинит и другие изменения сетчатки (93%)
ожирение (90%)
умственная отсталость (87%)
полидактилия обнаруживается у 60—70% больных
дефекты развития почек

Патогенез
Синдром Барде-Бидля принято считать аутосомно-рецесивным заболеванием, однако доказано, что для клинической
Патогенез
Синдром Барде-Бидля принято считать аутосомно-рецесивным заболеванием, однако доказано, что для клинической
Синдром Мак-Кьюна— Олбрайта— Брайцева (синдром МОБ, McCune–Albright–Braitsev syndrome) или синдром Олбрайта— Мак-Кьюна—
Синдром Мак-Кьюна— Олбрайта— Брайцева (синдром МОБ, McCune–Albright–Braitsev syndrome) или синдром Олбрайта— Мак-Кьюна—
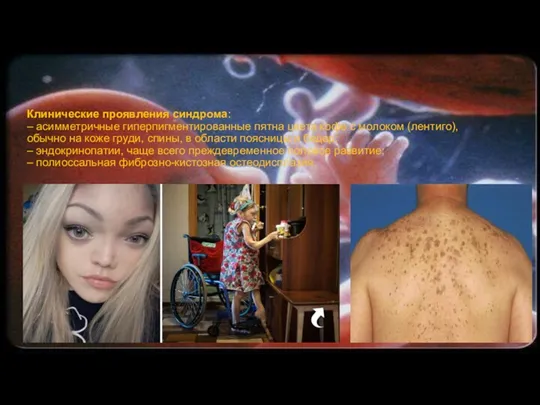
Клинические проявления синдрома:
– асимметричные гиперпигментированные пятна цвета кофе с молоком (лентиго),
Клинические проявления синдрома: – асимметричные гиперпигментированные пятна цвета кофе с молоком (лентиго),
Патогенез
Синдром МОБ обусловлен соматической мутацией гена GNASI, расположенного на длинном плече
Патогенез
Синдром МОБ обусловлен соматической мутацией гена GNASI, расположенного на длинном плече
Течение и прогноз
Заболевание клинически неоднородное. Наряду со случаями классической триады признаков, существуют
Течение и прогноз
Заболевание клинически неоднородное. Наряду со случаями классической триады признаков, существуют
 Компенсаторно-приспособительные реакции организма
Компенсаторно-приспособительные реакции организма Хирургическая (раневая) инфекция
Хирургическая (раневая) инфекция Кожный лейшманиоз
Кожный лейшманиоз Ерлер жыныс мүшелерінің жарақаттары
Ерлер жыныс мүшелерінің жарақаттары Дыхательная гимнастика
Дыхательная гимнастика Дәрігердің жұмысындағы қарым-қатынас кедергілері
Дәрігердің жұмысындағы қарым-қатынас кедергілері Профилактические программы, направленные на ограничение распространения ВИЧинфекции в мегаполисе
Профилактические программы, направленные на ограничение распространения ВИЧинфекции в мегаполисе Оценка качества препаратов, эффективности и безопасности профилактических и лечебных мероприятий
Оценка качества препаратов, эффективности и безопасности профилактических и лечебных мероприятий Отчет по направлению Trauma за 2019 год
Отчет по направлению Trauma за 2019 год Дошкольный и преддошкольный возраст
Дошкольный и преддошкольный возраст Токсикология тяжелых металлов
Токсикология тяжелых металлов Расстройства влечения
Расстройства влечения Дифтерия, көкжөтел, менингококк инфекцияларының этиологиясы және эпидемиологиялық шаралар
Дифтерия, көкжөтел, менингококк инфекцияларының этиологиясы және эпидемиологиялық шаралар Азбука электрокардиограммы
Азбука электрокардиограммы Медицина в России XIX века. Развитие отечественной хирургии. (Лекция 6)
Медицина в России XIX века. Развитие отечественной хирургии. (Лекция 6) Технические методы диагностических исследований и лечебных воздействий
Технические методы диагностических исследований и лечебных воздействий Болезнь Боткина. Гепатит А
Болезнь Боткина. Гепатит А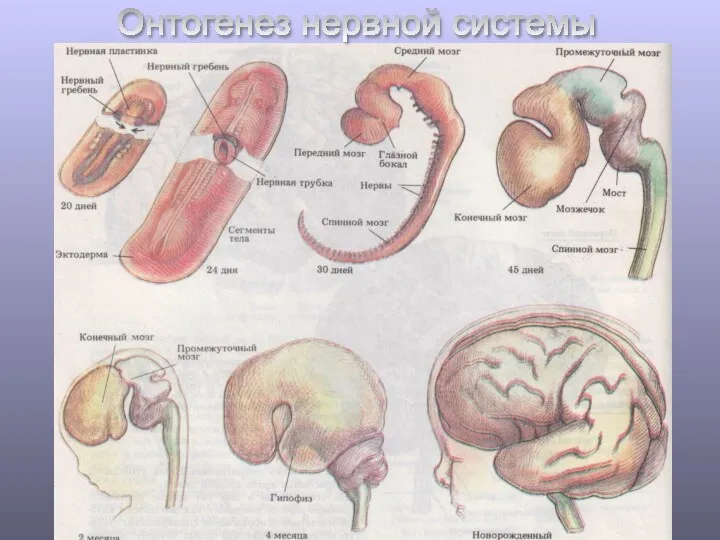 Онтогенез нервной системы. Анатомические обозначения
Онтогенез нервной системы. Анатомические обозначения Иммунитет и проблемы питания жителей современного города
Иммунитет и проблемы питания жителей современного города Организация санитарно-противоэпидемического обеспечения и медицинского снабжения в чрезвычайных ситуациях
Организация санитарно-противоэпидемического обеспечения и медицинского снабжения в чрезвычайных ситуациях Организация службы охраны репродуктивного здоровья девочек от 0 до 18 лет
Организация службы охраны репродуктивного здоровья девочек от 0 до 18 лет Электротравма
Электротравма Американская кардиологическая ассоциация (AHA)
Американская кардиологическая ассоциация (AHA) Харчова алергія у дітей з позиції доказової медицини
Харчова алергія у дітей з позиції доказової медицини Мейіргердің функциялық міндеттері
Мейіргердің функциялық міндеттері Пролежни. Лечение пролежней
Пролежни. Лечение пролежней Кровотечение и кровопотеря
Кровотечение и кровопотеря Зубы. Твердые и мягкие ткани зуба. Поддерживающий аппарат зуба
Зубы. Твердые и мягкие ткани зуба. Поддерживающий аппарат зуба