- Гепатолентикулярная дегенерация (Болезнь Вильсона-Коновалова)

Содержание
- 2. Болезнь Вильсона-Коновалова (ВК) - наследственное заболевание в основе которого лежит нарушение метаболизма меди и накопление ее
- 3. Накопление меди во внутренних органах Распространенность в различных регионах мира в среднем 1:30000
- 4. ВК - Аутосомно-рецессивный тип наследования Нарушается синтез медь-транспортирующего белка (АТР7В, ответственного за внутриклеточный транспорт ионов меди
- 5. Патогенез ВК
- 6. Патогенез ВК: Отложение меди в печени; Связывание с эритроцитами – гемолитическая анемия; Почках; Роговой оболочке глаз;
- 7. Накопление меди в гепатоцитах
- 8. КОЛЬЦА КАЙЗЕРА-ФЛЕЙШЕРА - располагаются в месте соединения склеры и роговицы, желтовато-зелёное или зелено-коричневое окрашивание краёв роговицы
- 9. Классификация В соответствии с клинической картиной отчетливо выделяются 3 формы заболевания: протекающие с поражением 1) печени;
- 10. Формы гепатолентикулярной дегенерации (ГД) Н. В. Коновалов (1948)
- 11. Начало заболевания: Может дебютировать в детском, подростковом, юношеском, зрелом возрасте; Очень редко – дебют ВК в
- 12. Классификация поражений печений при ВК I - латентная с неспецифическими морфологическими изменениями печени или мелко- или
- 13. Клиническая картина Дебют - в типичных случаях (у 42% больных) представлен одним из вариантов поражения печени:
- 14. Диагностические критерии **Штульман Д. Р., Левин О. С., 2004; Левин О.С. 2005 Celia H Chang, 2006
- 15. Диагноз Анамнез и физикальное обследование Диагностические тесты Развернутый анализ крови Сывороточный церулоплазмин Осмотр в щелевой лампе
- 16. Диагноз У больных с клинически латентным течением диагноз ставят при выявлении характерных изменений показателей метаболизма меди:
- 17. Церулоплазмин Сывороточный церулоплазмин должен рутинно определяться при обследовании больных с неустановленной печеночной, неврологической патологией и психиатрическим
- 18. Экскреция меди с мочой Для диагностики - базальное 24-часовое исследование мочи. Суточная экскреция мочи при БВ
- 19. Заподозрить раннюю стадию ВК можно на основании следующих признаков: Перенесенная желтуха Повторные кровотечения из носа, десен
- 21. Дифференциальный диагноз В зависимости от особенностей клинической картины БВ дифференциальный диагноз следует проводить: 1) с острыми
- 22. Если своевременно не начать лечение, направленное на выведение токсичных избытков меди из организма, то через 5–7
- 23. Лечение Идеально начинать лечение в «досимптомной стадии» ВК; Необходимо тщательное обследование всех братьев и сестер, если
- 24. Лечение: 1. Строгое соблюдение “печеночной” диеты (стол 5а): исключение богатых медью продуктов (шоколад, кофе, орехи, бобовые
- 25. Лечение Начальная доза D-пеницилламина - 0,25-0,5г/день с постепенным повышением (каждые 7 дней на 0,25 г) до
- 26. если к этому времени экскреция меди не превышает 150мкг/сут, после достижения клинического улучшения, которое обычно наступает
- 27. Оценку эффективности лечения следует производить не ранее, чем через 2 года от начала терапии; Недопустимы перерывы
- 28. Триентин Дозы: Внутрь натощак Дети Дети > 12 лет и взрослые: 750—1250 мг/сут в 2—4 приема;
- 29. Цинк - содержащие препараты* 25 мг – 3 - 4 раза в день *Sinha S; Taly
- 33. Мультисистемная атрофия спорадическая мультисистемная дегенерация преимущественно вовлекает базальные ганглии, оливы, мост, мозжечок, боковые рога спинного мозга,
- 34. Мультисистемная атрофия Критерии диагностики: - Вегетативная/тазовая дисфункция (ортостатическая гипотензия со снижение систолического АД не менее чем
- 35. Мозжечковая атаксия (статолокомоторная атаксия в сочетании с не менее чем одним другим мозжечковым симптомом - дизартрией,
- 36. Критерии, исключающие диагноз: - Начало в возрасте до 30 лет. - Положительный семейный анамнез. - Наличие
- 37. Болезнь Гентингтона наследственное нейродегенеративное заболевание с выраженной психопатологической симптоматикой - имеет аутосомно-доминантный тип наследования со 100%-й
- 38. Болезнь Гентингтона
- 39. Эпидемиология: 5-8/100.000 населения Клиника: Обычно заболевание начинается в возрасте 30- 40 лет; типичная продолжительность - 17
- 40. Двигательные расстройства начинаются с нарушений координации, стереотипных мелких движений пальцев, непроизвольного гримасничанья, хаотического разбрасывания рук и
- 41. Деменция; Депрессия; Суициды; Психозы.
- 43. Скачать презентацию

Болезнь Вильсона-Коновалова (ВК)
- наследственное заболевание в основе которого лежит нарушение метаболизма
Болезнь Вильсона-Коновалова (ВК)
- наследственное заболевание в основе которого лежит нарушение метаболизма
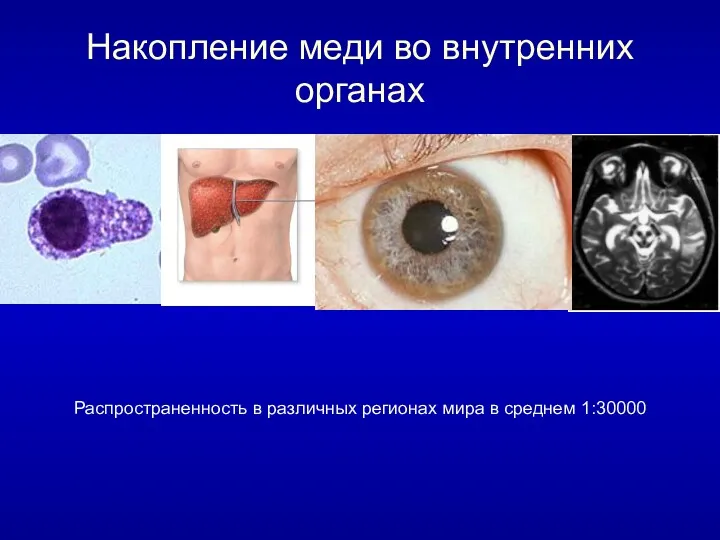
Накопление меди во внутренних органах
Распространенность в различных регионах мира в
Накопление меди во внутренних органах
Распространенность в различных регионах мира в

ВК - Аутосомно-рецессивный тип наследования
Нарушается синтез медь-транспортирующего белка (АТР7В, ответственного за
ВК - Аутосомно-рецессивный тип наследования
Нарушается синтез медь-транспортирующего белка (АТР7В, ответственного за
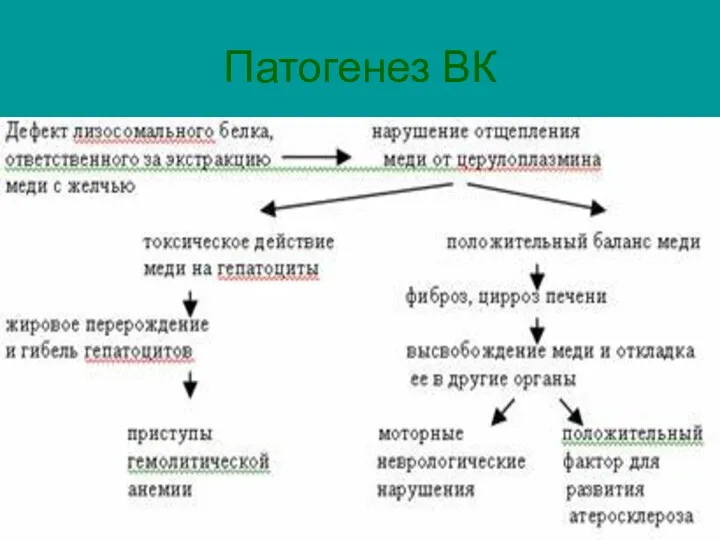
Патогенез ВК
Патогенез ВК

Патогенез ВК:
Отложение меди в печени;
Связывание с эритроцитами – гемолитическая анемия;
Почках;
Роговой оболочке
Патогенез ВК:
Отложение меди в печени;
Связывание с эритроцитами – гемолитическая анемия;
Почках;
Роговой оболочке
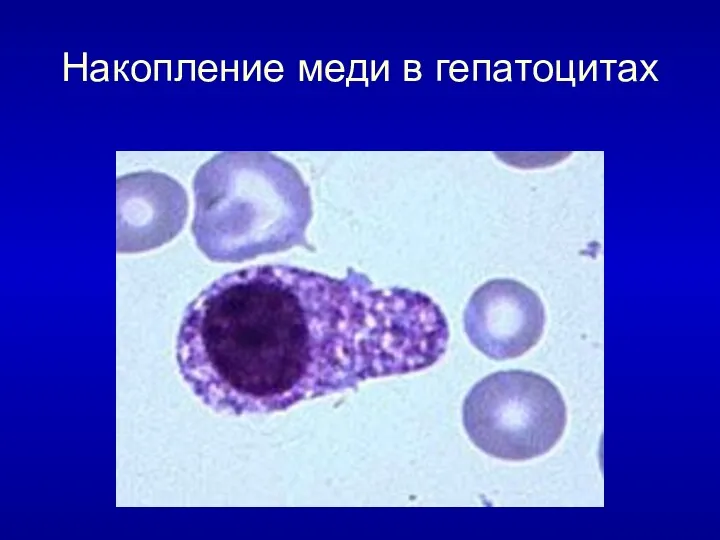
Накопление меди в гепатоцитах
Накопление меди в гепатоцитах
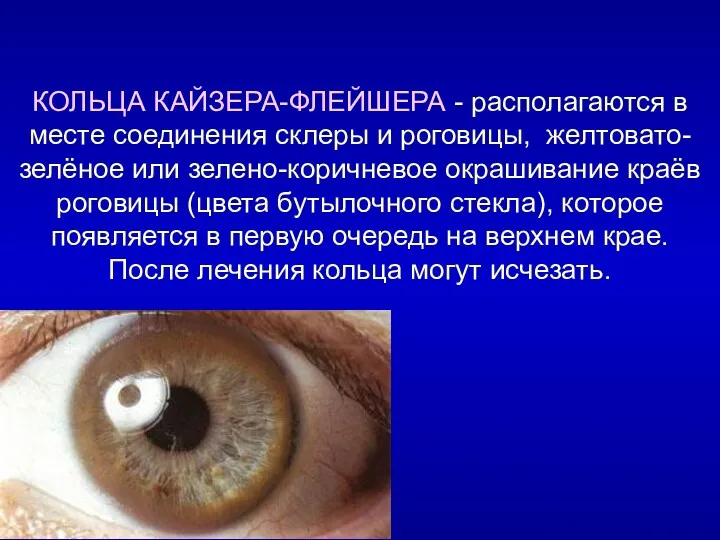
КОЛЬЦА КАЙЗЕРА-ФЛЕЙШЕРА - располагаются в месте соединения склеры и роговицы, желтовато-зелёное
КОЛЬЦА КАЙЗЕРА-ФЛЕЙШЕРА - располагаются в месте соединения склеры и роговицы, желтовато-зелёное

Классификация
В соответствии с клинической картиной отчетливо выделяются 3 формы заболевания:
протекающие
Классификация
В соответствии с клинической картиной отчетливо выделяются 3 формы заболевания:
протекающие

Формы гепатолентикулярной дегенерации (ГД)
Н. В. Коновалов (1948)
Формы гепатолентикулярной дегенерации (ГД)
Н. В. Коновалов (1948)

Начало заболевания:
Может дебютировать в детском, подростковом, юношеском, зрелом возрасте;
Очень редко –
Начало заболевания:
Может дебютировать в детском, подростковом, юношеском, зрелом возрасте;
Очень редко –

Классификация поражений печений при ВК
I - латентная с неспецифическими морфологическими изменениями печени
Классификация поражений печений при ВК
I - латентная с неспецифическими морфологическими изменениями печени

Клиническая картина
Дебют - в типичных случаях (у 42% больных) представлен одним
Клиническая картина
Дебют - в типичных случаях (у 42% больных) представлен одним

Диагностические критерии
**Штульман Д. Р., Левин О. С., 2004; Левин О.С. 2005
Диагностические критерии
**Штульман Д. Р., Левин О. С., 2004; Левин О.С. 2005

Диагноз
Анамнез и физикальное обследование
Диагностические тесты
Развернутый анализ крови
Сывороточный
Диагноз
Анамнез и физикальное обследование
Диагностические тесты
Развернутый анализ крови
Сывороточный

Диагноз
У больных с клинически латентным течением диагноз ставят при выявлении характерных
Диагноз
У больных с клинически латентным течением диагноз ставят при выявлении характерных

Церулоплазмин
Сывороточный церулоплазмин должен рутинно определяться при обследовании больных с неустановленной
Церулоплазмин
Сывороточный церулоплазмин должен рутинно определяться при обследовании больных с неустановленной

Экскреция меди с мочой
Для диагностики - базальное 24-часовое исследование мочи.
Экскреция меди с мочой
Для диагностики - базальное 24-часовое исследование мочи.

Заподозрить раннюю стадию ВК можно на основании следующих признаков:
Перенесенная желтуха
Повторные кровотечения
Заподозрить раннюю стадию ВК можно на основании следующих признаков:
Перенесенная желтуха
Повторные кровотечения


Дифференциальный диагноз
В зависимости от особенностей клинической картины БВ дифференциальный диагноз следует
Дифференциальный диагноз
В зависимости от особенностей клинической картины БВ дифференциальный диагноз следует

Если своевременно не начать лечение, направленное на выведение токсичных избытков меди
Если своевременно не начать лечение, направленное на выведение токсичных избытков меди

Лечение
Идеально начинать лечение в «досимптомной стадии» ВК;
Необходимо тщательное обследование всех братьев
Лечение
Идеально начинать лечение в «досимптомной стадии» ВК;
Необходимо тщательное обследование всех братьев

Лечение:
1. Строгое соблюдение “печеночной” диеты (стол 5а): исключение богатых медью продуктов
Лечение:
1. Строгое соблюдение “печеночной” диеты (стол 5а): исключение богатых медью продуктов

Лечение
Начальная доза D-пеницилламина - 0,25-0,5г/день с постепенным повышением (каждые 7
Лечение
Начальная доза D-пеницилламина - 0,25-0,5г/день с постепенным повышением (каждые 7

если к этому времени экскреция меди не превышает 150мкг/сут, после достижения
если к этому времени экскреция меди не превышает 150мкг/сут, после достижения

Оценку эффективности лечения следует производить не ранее, чем через 2 года
Оценку эффективности лечения следует производить не ранее, чем через 2 года

Триентин
Дозы:
Внутрь натощак
Дети < 12 лет: 500—750 мг/сут в 2—4 приема, максимальная доза 1,5 г/сут.
Дети > 12 лет и
Триентин
Дозы:
Внутрь натощак
Дети < 12 лет: 500—750 мг/сут в 2—4 приема, максимальная доза 1,5 г/сут.
Дети > 12 лет и

Цинк - содержащие препараты*
25 мг – 3 - 4 раза в
Цинк - содержащие препараты*
25 мг – 3 - 4 раза в

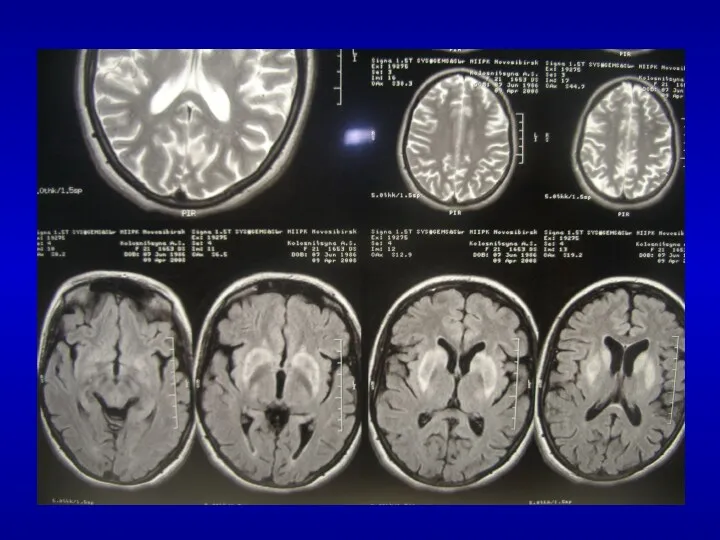
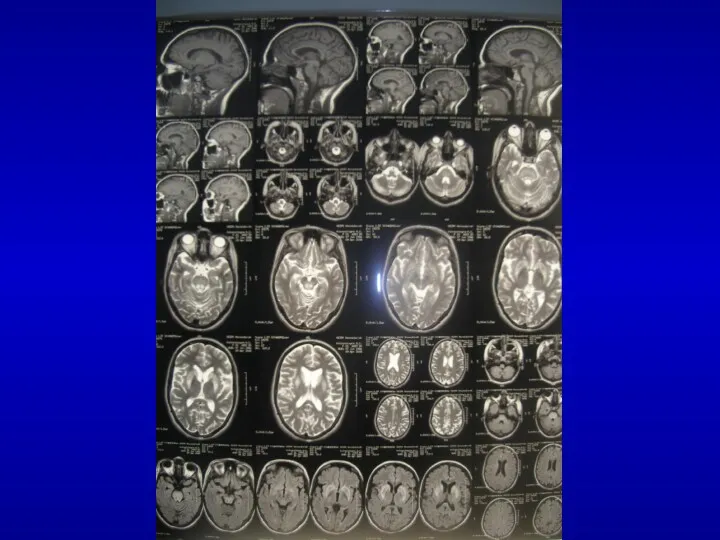

Мультисистемная атрофия
спорадическая мультисистемная дегенерация
преимущественно вовлекает базальные ганглии, оливы, мост, мозжечок, боковые
Мультисистемная атрофия
спорадическая мультисистемная дегенерация
преимущественно вовлекает базальные ганглии, оливы, мост, мозжечок, боковые

Мультисистемная атрофия
Критерии диагностики:
- Вегетативная/тазовая дисфункция (ортостатическая гипотензия со снижение систолического
Мультисистемная атрофия
Критерии диагностики:
- Вегетативная/тазовая дисфункция (ортостатическая гипотензия со снижение систолического

Мозжечковая атаксия (статолокомоторная атаксия в сочетании с не менее чем одним
Мозжечковая атаксия (статолокомоторная атаксия в сочетании с не менее чем одним

Критерии, исключающие диагноз:
- Начало в возрасте до 30 лет.
- Положительный семейный
Критерии, исключающие диагноз:
- Начало в возрасте до 30 лет.
- Положительный семейный

Болезнь Гентингтона
наследственное нейродегенеративное заболевание с выраженной психопатологической симптоматикой - имеет аутосомно-доминантный
Болезнь Гентингтона
наследственное нейродегенеративное заболевание с выраженной психопатологической симптоматикой - имеет аутосомно-доминантный
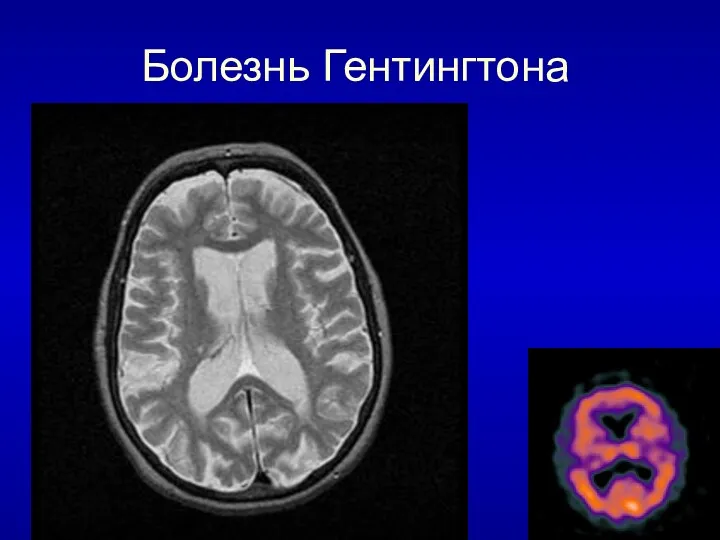
Болезнь Гентингтона
Болезнь Гентингтона

Эпидемиология: 5-8/100.000 населения
Клиника:
Обычно заболевание начинается в возрасте 30- 40 лет;
типичная
Эпидемиология: 5-8/100.000 населения
Клиника:
Обычно заболевание начинается в возрасте 30- 40 лет;
типичная

Двигательные расстройства
начинаются с нарушений координации, стереотипных мелких движений пальцев, непроизвольного гримасничанья,
Двигательные расстройства
начинаются с нарушений координации, стереотипных мелких движений пальцев, непроизвольного гримасничанья,

Деменция;
Депрессия;
Суициды;
Психозы.
Деменция;
Депрессия;
Суициды;
Психозы.
 Клинический случай. Диарейный синдром
Клинический случай. Диарейный синдром Диагностика вирусных инфекций
Диагностика вирусных инфекций Менінгококова інфекція
Менінгококова інфекція Туберкулез внутригрудных лимфатических узлов
Туберкулез внутригрудных лимфатических узлов Дифференциальная диагностика менингеального синдрома
Дифференциальная диагностика менингеального синдрома Тромбоемболія легеневої артерії (ТЕЛА)
Тромбоемболія легеневої артерії (ТЕЛА) Первая медицинская помощь при острых отравлениях
Первая медицинская помощь при острых отравлениях Балалардағы бет – жақ аймағындағы ауытқуларды емдеуде миогимнастикасы
Балалардағы бет – жақ аймағындағы ауытқуларды емдеуде миогимнастикасы Профилактика инсультов у пациентов с фибрилляцией предсердий
Профилактика инсультов у пациентов с фибрилляцией предсердий Регуляция иммунного ответа. Цитокины, факторы роста, гормоны. (Лекция 4)
Регуляция иммунного ответа. Цитокины, факторы роста, гормоны. (Лекция 4) Стоматологическое материаловедение
Стоматологическое материаловедение Синдромы функциональной несформированности, дефицитарности отделов головного мозга
Синдромы функциональной несформированности, дефицитарности отделов головного мозга Злокачественные опухоли и их профилактика
Злокачественные опухоли и их профилактика Инсулинотерапия при СД 2 типа
Инсулинотерапия при СД 2 типа Болезнь Дюринга (герпетифорный дерматит), многоформной экссудативной эритеме, синдроме Стивенса-Джонсона
Болезнь Дюринга (герпетифорный дерматит), многоформной экссудативной эритеме, синдроме Стивенса-Джонсона Организационные вопросы онкологической помощи в Российской Федерации
Организационные вопросы онкологической помощи в Российской Федерации Жоғарғы жүйке жүйесі бұзылыстарының балалардағы ерекшеліктері
Жоғарғы жүйке жүйесі бұзылыстарының балалардағы ерекшеліктері Методы лучевой диагностики
Методы лучевой диагностики Туберкулез. Микобактериозы
Туберкулез. Микобактериозы Активное ведение третьего периода родов
Активное ведение третьего периода родов Многоплодная беременность при экстракорпоральном оплодотворении
Многоплодная беременность при экстракорпоральном оплодотворении Болезнь Вакеза. Эритремия: критерии диагноза, этиология, патогенез, клиника, лечение
Болезнь Вакеза. Эритремия: критерии диагноза, этиология, патогенез, клиника, лечение Виписування, зберігання та застосування ліків
Виписування, зберігання та застосування ліків Этика и деонтология в педиатрии. Периоды детского возраста. Особенности ухода за детьми различного возраста
Этика и деонтология в педиатрии. Периоды детского возраста. Особенности ухода за детьми различного возраста Инфаркт миокарда. Роль медицинской сестры в лечении и профилактике
Инфаркт миокарда. Роль медицинской сестры в лечении и профилактике Адгезивті және алмалы көпіртәрізді протездермен, протездеу
Адгезивті және алмалы көпіртәрізді протездермен, протездеу БАБЖ бағдарламасы. Жөтел нмесе тыныс алудың қиындауы бар науқастарды жұргізу тәсілі
БАБЖ бағдарламасы. Жөтел нмесе тыныс алудың қиындауы бар науқастарды жұргізу тәсілі ЗОЖ (2). Развитие способностей. Задание на лето
ЗОЖ (2). Развитие способностей. Задание на лето