- Хронические лейкозы

Содержание
- 2. Схема гемопоэза Стволовая клетка крови Клетка предшественница миелопоэза Клетка предшественница лимфопоэза лейкопоэтин эритропоэтин тромбопоэтин миелобласт эритробласт
- 3. Схема гемопоэза Клетка предшественница лимфопоэза Клетка предшественница Т-лимфоцитов Клетка предшественница В-лимфоцитов Т-лимфобласт Т-пролимфоцит Т-лимфоцит Т-иммунобласт Активированный
- 4. Хронические лейкозы -опухоли кроветворной ткани, основной субстрат которых составляют созревающие и зрелые клетки
- 5. Хронические лейкозы При хроническом лейкозе опухолевые клетки напоминают нормальные, но отличаются от них. Они живут слишком
- 6. Этиология Этиология лейкозов до настоящего времени точно не установлена. Об опухолевой природе лейкозов свидетельствует наличие общих
- 8. Миелопролиферативные Лимфопролиферативные Хронический миелолейкоз Эритремия Хронический мегакариоцитарный лейкоз Идиопатический миелофиброз Болезнь тяжелых цепей Болезнь Вальденстрема Миеломная
- 9. Хронический миелолейкоз ХМЛ - хроническое миелопролиферативное заболевание, при котором отмечают усиление образования гранулоцитов (преимущественно нейтрофилов, а
- 11. Этиология и патогенез Причина патологического роста клеток - мутация клетки-предшественницы миелопоэза (частично детерминированная полипотентная клетка). Это
- 12. Стадии хронического миелолейкоза ХМЛ в своем развитии закономерно проходит две стадии - моноклоновую и поликлоновую, которым
- 13. Хроническая фаза Миелоидная пролиферация костного мозга и небольшие изменения в крови (до 1-3% бластов) без признаков
- 14. Фаза акселерации Выраженные клинико-гематологические нарушения (интоксикация продуктами распада лейкозных клеток, увеличение печени и селезенки, миелоидная пролиферация
- 15. Бластный криз Развитие поликлоновой опухоли: рефрактерность к проводимому цитостатическому лечению, истощение, значительное увеличение селезенки и печени,
- 16. Клиническая картина • Миелопролиферативный синдром, в основе которого лежит миелоидная пролиферация костного мозга, включает: - общие
- 17. Диагностика • лейкоцитоз более 20х109/л; • присутствие в лейкоцитарной формуле пролиферирующих форм (миелобластов и промиелоцитов) и
- 18. Диагностика I стадия: периферической крови обнаруживают лейкоцитоз (более 50х109/л) с нейтрофилезом, гранулоциты на всех стадиях созревания
- 19. Лечение ХМЛ Иматиниб (гливек) – специфически ингибирует BCR-ABL-тирозинкиназную активность и подавляет бесконтрольную пролиферацию лейкоцитов. У 76%
- 20. Методы терапии ХМЛ Выбор метода терапии в ранней хронической стадии ХМЛ I. Нетрансплантанционные методы (расположены в
- 21. Аллотрансплантация костного мозга? До открытия иматиниба ТКМ считали единственным радикальным лечением заболевания. Существенная роль ТКМ остается
- 22. Эритремия (истинная полицитемия, болезнь Вакеза) Миелопролиферативное заболевание, хронический, доброкачественно текущий лейкоз, при котором отмечают усиленное образование
- 23. Картина крови Увеличиваются эритроциты, Нв, гематокрит, вязкость. СОЭ –резко замедлено. В последующем присоединяются лейкоцитоз, нейтрофилез, иногда
- 24. Клиника Плеторический синдром обусловлен увеличенным содержанием эритроцитов, лейкоцитов и тромбоцитов: • из субъективных синдромов (головную боль,
- 25. Клиника Миелопролиферативный синдром обусловлен гиперплазией всех трех ростков кроветворения: • субъективные симптомы (слабость, потливость, повышение температуры
- 27. Лечение эритремии В начальной стадии могут быть эффективны кровопускания – по 500 мл через день до
- 28. Осложнения • сосудистый тромбоз (мозговых, коронарных, периферических артерий); • геморрагический синдром (кровотечения после малых оперативных вмешательств
- 29. Хронический идиопатический миелофиброз Усиленная пролиферация (размножение) клеток костного мозга (КМ) с последующим прогрессированием в миелофиброз (фиброз
- 30. Клиника В начальной стадии 1/3 пациентов жалоб не имеют. ИМФ подозревают при обнаружении спленомегалии Повышение тромбоцитов
- 31. Диагноз Для постановке диагноза необходимо наличие всех 3-х основных и 2-х малых критериев Основные (большие) критерии
- 32. Исследование крови Нормоцитарная анемия у 54% пациентов Количество тромбоцитов и лейкоцитов повышено в начальной стадии ХИМ
- 33. Морфология клеток КМ Гиперклеточный КМ с признаками гиперплазии всех 3-х клеточных линий в начальной стадии ХИМ
- 34. Лечение Симптоматическая терапия В некоторых случаях – аллогенная трансплантация стволовых клеток Иногда ингибитор JAK2, руксолитиниб
- 35. Геморрагическая тромбоцитемия (хр. мегакариоцитарный лейкоз) редкий гемобластоз , возникающий на уровне стволовой клетки. Заболеваемость проявляется беспричинным
- 36. Основные характеристики Гипертромбоцитозом в крови, иногда 3— 4 млн в 1 мкл Лейкоцитоза, сдвига лейкоцитарной формулы
- 37. Хронический лимфолейкоз Хронический лимфолейкоз — В- клеточное лимфопролиферативное неопластическое заболевание, характеризующееся пролиферацией и увеличением в периферической
- 38. Подобные лимфоциты функционально неполноценны, что выражается в нарушении функций иммунной системы, повышенной склонности к аутоиммунным реакциям
- 39. Распространенность ХЛЛ - одна из самых частых разновидностей лейкозов (30% всех случаев). В 95% случаев ХЛЛ
- 40. Этиология • Вирусная инфекция (Эпштейн-Барра) • Хромосомные аномалии в области 12, 13, 14-й хромосом. • Самая
- 41. Патогенез • отсутствуют признаки опухолевой прогрессии (бластный криз очень редко возникает в терминальной фазе); • нет
- 42. Классификация ХЛЛ (по K.R. Rai, 1975) • 0 - абсолютный лимфоцитоз без видимого увеличения лимфатических узлов
- 43. Классификация ХЛЛ (по J.L. Binet, 1977) Стадия : А(выживаемость > 10 лет) Лимфоцитоз + вовлечение менее
- 44. В зависимости от клинической картины • опухолевый - периферические лимфатические узлы резко увеличены, плотные, малоподвижные, резко
- 45. Клиника • Лимфопролиферативный, обусловленный лимфаденопатией, спленомегалией и лимфоидной пролиферацией костного мозга: - общие симптомы, обусловленные интоксикацией
- 46. Клиника • Синдром осложнений: - гнойно-воспалительных; - аутоиммунных (аутоиммунная гемолитическая анемия). Различная выраженность синдромов на тех
- 47. Диагностика В общем анализе крови: • количество лейкоцитов от 50х109/л до 100 х 109/л и даже
- 49. Примерно у 50% больных ХЛЛ обнаруживается нормохромная нормоцитарная анемия. Этиология: • лейкозная инфильтрация костного мозга (обычно
- 50. Диагностика 2) Биохимический анализ крови: определение содержания белка, белковых фракций, билирубина, аминотрансфераз, щелочной фосфатазы, глюкоза, железа,
- 51. Диагностика 4) Иммунофенотипирование лимфоцитов!!!! – ведущий метод диагностика – определение – CD- кластеров на поверхности лимфоцитов
- 52. Стернальная пункция Выраженная лимфоидная инфильтрация. Лимфоциты составляют более 30% (иногда 50-60% и даже больше) от общего
- 53. Лечение • режим • цитостатическая терапия • лечебный лимфоцитоферез • лучевая терапия • спленэктомия • ГКС
- 54. Показания к цитостатической терапии 1) Появление симптомов интоксикации; 2) Появление анемии или тромбоцитопении; 3) Нарастание лимфоаденопатии,
- 55. Современные подходы к терапии ХЛЛ • Ритуксимаб (Мабтера) - моноклональное антитело к антигену СD 20. Вызывает:
- 58. Скачать презентацию
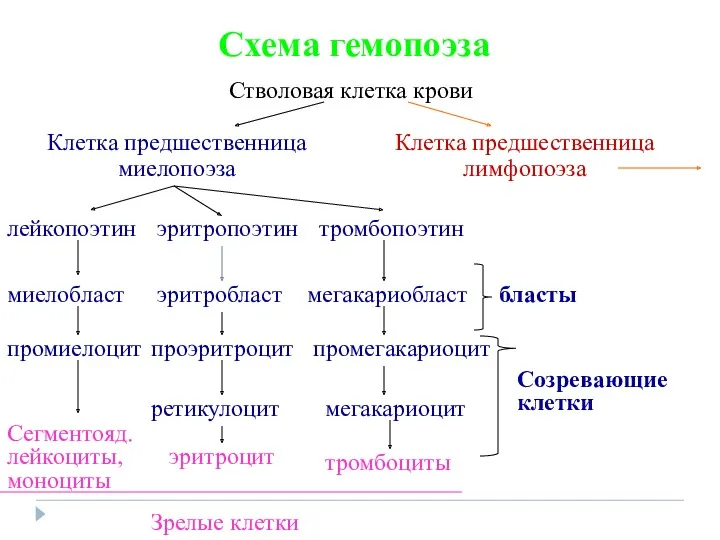
Схема гемопоэза
Стволовая клетка крови
Клетка предшественница миелопоэза
Клетка предшественница лимфопоэза
лейкопоэтин
эритропоэтин
тромбопоэтин
миелобласт
эритробласт
мегакариобласт
промиелоцит
проэритроцит
промегакариоцит
Сегментояд. лейкоциты, моноциты
ретикулоцит
эритроцит
мегакариоцит
тромбоциты
Зрелые клетки
бласты
Созревающие
Схема гемопоэза
Стволовая клетка крови
Клетка предшественница миелопоэза
Клетка предшественница лимфопоэза
лейкопоэтин
эритропоэтин
тромбопоэтин
миелобласт
эритробласт
мегакариобласт
промиелоцит
проэритроцит
промегакариоцит
Сегментояд. лейкоциты, моноциты
ретикулоцит
эритроцит
мегакариоцит
тромбоциты
Зрелые клетки
бласты
Созревающие
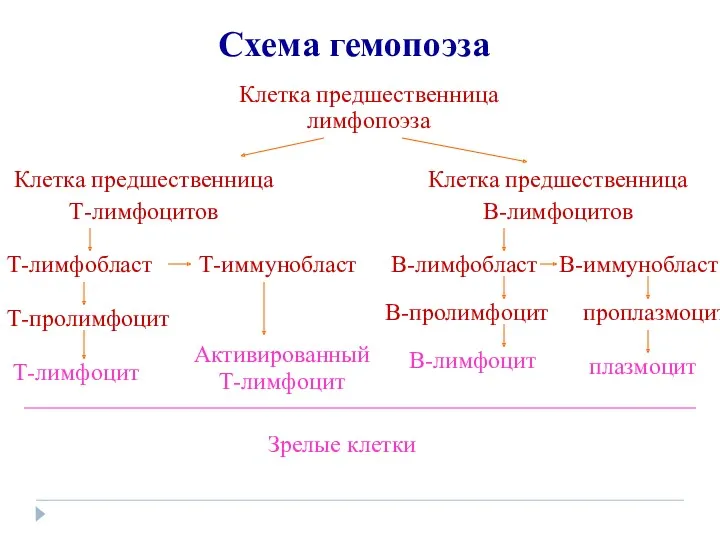
Схема гемопоэза
Клетка предшественница лимфопоэза
Клетка предшественница
Т-лимфоцитов
Клетка предшественница
В-лимфоцитов
Т-лимфобласт
Т-пролимфоцит
Т-лимфоцит
Т-иммунобласт
Активированный
Т-лимфоцит
В-лимфобласт
В-пролимфоцит
В-лимфоцит
В-иммунобласт
проплазмоцит
плазмоцит
Зрелые клетки
Схема гемопоэза
Клетка предшественница лимфопоэза
Клетка предшественница
Т-лимфоцитов
Клетка предшественница
В-лимфоцитов
Т-лимфобласт
Т-пролимфоцит
Т-лимфоцит
Т-иммунобласт
Активированный
Т-лимфоцит
В-лимфобласт
В-пролимфоцит
В-лимфоцит
В-иммунобласт
проплазмоцит
плазмоцит
Зрелые клетки
Хронические лейкозы
-опухоли кроветворной ткани, основной субстрат которых составляют созревающие и зрелые
Хронические лейкозы
-опухоли кроветворной ткани, основной субстрат которых составляют созревающие и зрелые

Хронические лейкозы
При хроническом лейкозе опухолевые клетки напоминают нормальные, но отличаются
Хронические лейкозы
При хроническом лейкозе опухолевые клетки напоминают нормальные, но отличаются

Этиология
Этиология лейкозов до настоящего времени точно не установлена. Об опухолевой природе
Этиология
Этиология лейкозов до настоящего времени точно не установлена. Об опухолевой природе
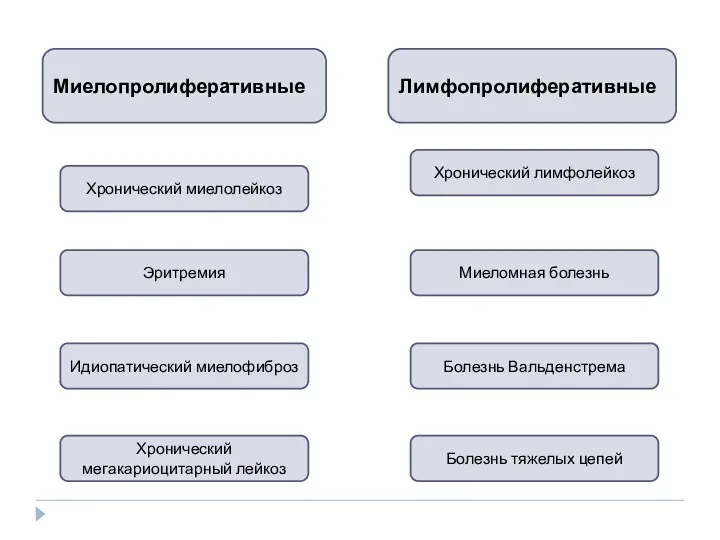
Миелопролиферативные
Лимфопролиферативные
Хронический миелолейкоз
Эритремия
Хронический мегакариоцитарный лейкоз
Идиопатический миелофиброз
Болезнь тяжелых цепей
Болезнь Вальденстрема
Миеломная болезнь
Хронический лимфолейкоз
Миелопролиферативные
Лимфопролиферативные
Хронический миелолейкоз
Эритремия
Хронический мегакариоцитарный лейкоз
Идиопатический миелофиброз
Болезнь тяжелых цепей
Болезнь Вальденстрема
Миеломная болезнь
Хронический лимфолейкоз

Хронический миелолейкоз
ХМЛ - хроническое миелопролиферативное заболевание, при котором отмечают усиление образования
Хронический миелолейкоз
ХМЛ - хроническое миелопролиферативное заболевание, при котором отмечают усиление образования


Этиология и патогенез
Причина патологического роста клеток - мутация клетки-предшественницы миелопоэза (частично
Этиология и патогенез
Причина патологического роста клеток - мутация клетки-предшественницы миелопоэза (частично

Стадии хронического миелолейкоза
ХМЛ в своем развитии закономерно проходит две стадии -
Стадии хронического миелолейкоза
ХМЛ в своем развитии закономерно проходит две стадии -
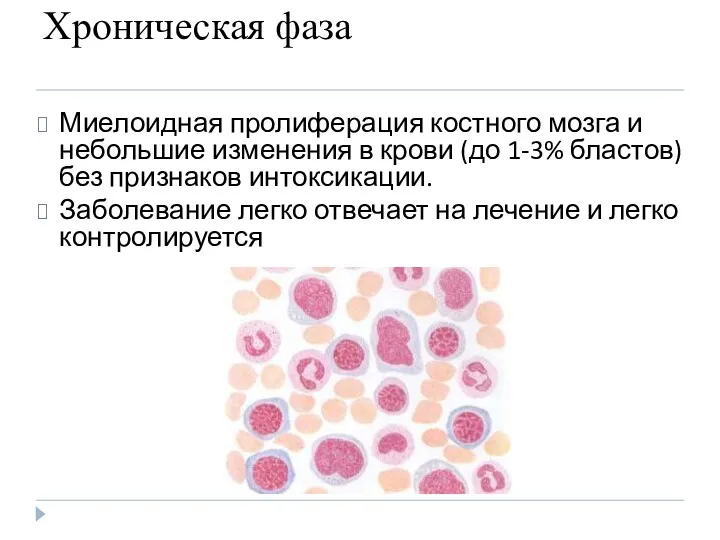
Хроническая фаза
Миелоидная пролиферация костного мозга и небольшие изменения в крови (до
Хроническая фаза
Миелоидная пролиферация костного мозга и небольшие изменения в крови (до

Фаза акселерации
Выраженные клинико-гематологические нарушения (интоксикация продуктами распада лейкозных клеток, увеличение печени
Фаза акселерации
Выраженные клинико-гематологические нарушения (интоксикация продуктами распада лейкозных клеток, увеличение печени

Бластный криз
Развитие поликлоновой опухоли: рефрактерность к проводимому цитостатическому лечению, истощение, значительное
Бластный криз
Развитие поликлоновой опухоли: рефрактерность к проводимому цитостатическому лечению, истощение, значительное

Клиническая картина
• Миелопролиферативный синдром, в основе которого лежит миелоидная пролиферация костного мозга,
Клиническая картина
• Миелопролиферативный синдром, в основе которого лежит миелоидная пролиферация костного мозга,

Диагностика
• лейкоцитоз более 20х109/л;
• присутствие в лейкоцитарной формуле пролиферирующих форм (миелобластов и промиелоцитов)
Диагностика
• лейкоцитоз более 20х109/л;
• присутствие в лейкоцитарной формуле пролиферирующих форм (миелобластов и промиелоцитов)

Диагностика
I стадия: периферической крови обнаруживают лейкоцитоз (более 50х109/л) с нейтрофилезом, гранулоциты
Диагностика
I стадия: периферической крови обнаруживают лейкоцитоз (более 50х109/л) с нейтрофилезом, гранулоциты

Лечение ХМЛ
Иматиниб (гливек) – специфически ингибирует BCR-ABL-тирозинкиназную активность и подавляет бесконтрольную
Лечение ХМЛ
Иматиниб (гливек) – специфически ингибирует BCR-ABL-тирозинкиназную активность и подавляет бесконтрольную

Методы терапии ХМЛ
Выбор метода терапии в ранней хронической стадии ХМЛ
I. Нетрансплантанционные
Методы терапии ХМЛ
Выбор метода терапии в ранней хронической стадии ХМЛ
I. Нетрансплантанционные

Аллотрансплантация костного мозга?
До открытия иматиниба ТКМ считали единственным радикальным лечением заболевания.
Аллотрансплантация костного мозга?
До открытия иматиниба ТКМ считали единственным радикальным лечением заболевания.
Эритремия (истинная полицитемия, болезнь Вакеза)
Миелопролиферативное заболевание, хронический, доброкачественно текущий лейкоз, при
Эритремия (истинная полицитемия, болезнь Вакеза)
Миелопролиферативное заболевание, хронический, доброкачественно текущий лейкоз, при

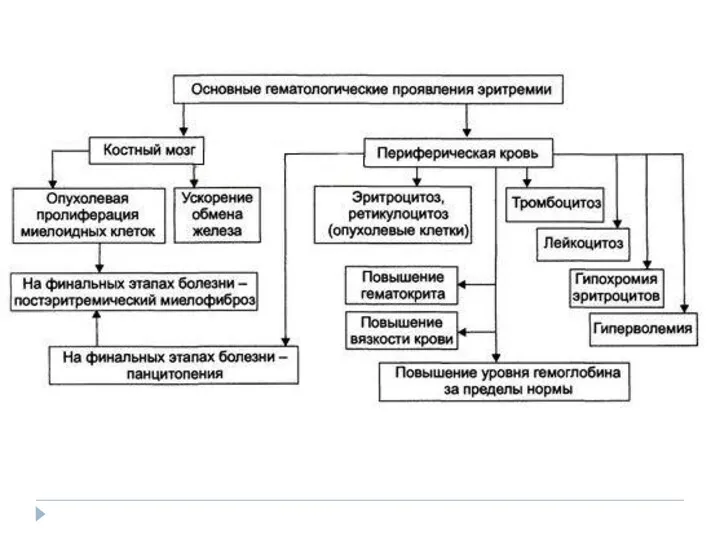
Картина крови
Увеличиваются эритроциты, Нв, гематокрит, вязкость. СОЭ –резко замедлено.
В последующем
Картина крови
Увеличиваются эритроциты, Нв, гематокрит, вязкость. СОЭ –резко замедлено.
В последующем

Клиника
Плеторический синдром обусловлен увеличенным содержанием эритроцитов, лейкоцитов и тромбоцитов:
• из субъективных
Клиника
Плеторический синдром обусловлен увеличенным содержанием эритроцитов, лейкоцитов и тромбоцитов:
• из субъективных

Клиника
Миелопролиферативный синдром обусловлен гиперплазией всех трех ростков кроветворения:
• субъективные симптомы (слабость,
Клиника
Миелопролиферативный синдром обусловлен гиперплазией всех трех ростков кроветворения:
• субъективные симптомы (слабость,

Лечение эритремии
В начальной стадии могут быть эффективны кровопускания – по 500
Лечение эритремии
В начальной стадии могут быть эффективны кровопускания – по 500

Осложнения
• сосудистый тромбоз (мозговых, коронарных, периферических артерий);
• геморрагический синдром (кровотечения после малых оперативных
Осложнения
• сосудистый тромбоз (мозговых, коронарных, периферических артерий);
• геморрагический синдром (кровотечения после малых оперативных

Хронический идиопатический миелофиброз
Усиленная пролиферация (размножение) клеток костного мозга (КМ) с последующим
Хронический идиопатический миелофиброз
Усиленная пролиферация (размножение) клеток костного мозга (КМ) с последующим

Клиника
В начальной стадии 1/3 пациентов жалоб не имеют. ИМФ подозревают при
Клиника
В начальной стадии 1/3 пациентов жалоб не имеют. ИМФ подозревают при

Диагноз
Для постановке диагноза необходимо наличие всех 3-х основных и 2-х малых
Диагноз
Для постановке диагноза необходимо наличие всех 3-х основных и 2-х малых
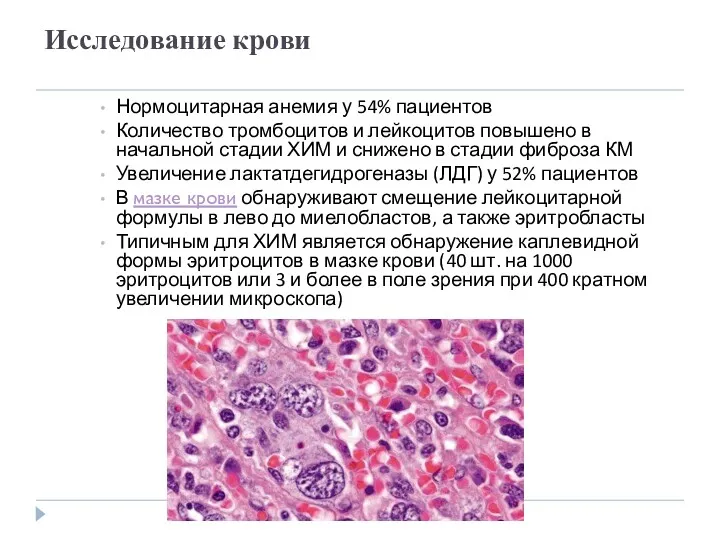
Исследование крови
Нормоцитарная анемия у 54% пациентов
Количество тромбоцитов и лейкоцитов повышено
Исследование крови
Нормоцитарная анемия у 54% пациентов
Количество тромбоцитов и лейкоцитов повышено

Морфология клеток КМ
Гиперклеточный КМ с признаками гиперплазии всех 3-х клеточных линий
Морфология клеток КМ
Гиперклеточный КМ с признаками гиперплазии всех 3-х клеточных линий

Лечение
Симптоматическая терапия
В некоторых случаях – аллогенная трансплантация стволовых клеток
Иногда ингибитор
Лечение
Симптоматическая терапия
В некоторых случаях – аллогенная трансплантация стволовых клеток
Иногда ингибитор

Геморрагическая тромбоцитемия (хр. мегакариоцитарный лейкоз)
редкий гемобластоз , возникающий на уровне стволовой
Геморрагическая тромбоцитемия (хр. мегакариоцитарный лейкоз)
редкий гемобластоз , возникающий на уровне стволовой

Основные характеристики
Гипертромбоцитозом в крови, иногда 3— 4 млн в 1 мкл
Лейкоцитоза,
Основные характеристики
Гипертромбоцитозом в крови, иногда 3— 4 млн в 1 мкл
Лейкоцитоза,

Хронический лимфолейкоз
Хронический лимфолейкоз — В- клеточное лимфопролиферативное неопластическое заболевание, характеризующееся пролиферацией
Хронический лимфолейкоз
Хронический лимфолейкоз — В- клеточное лимфопролиферативное неопластическое заболевание, характеризующееся пролиферацией
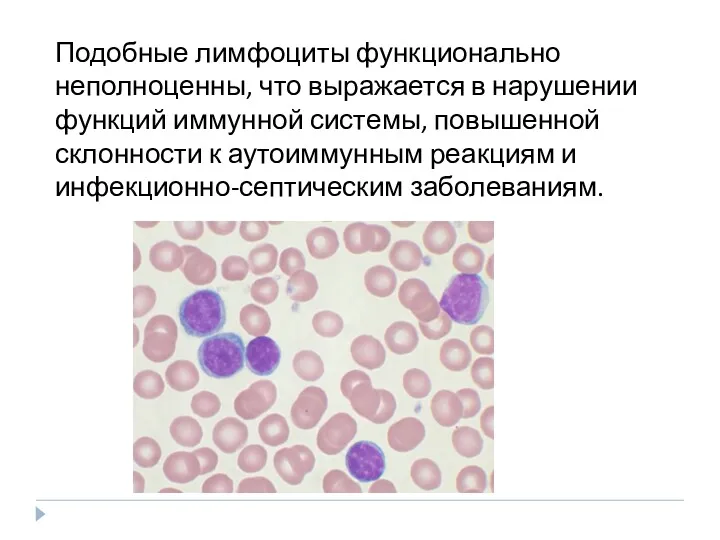
Подобные лимфоциты функционально неполноценны, что выражается в нарушении функций иммунной системы,
Подобные лимфоциты функционально неполноценны, что выражается в нарушении функций иммунной системы,

Распространенность
ХЛЛ - одна из самых частых разновидностей лейкозов (30% всех случаев).
Распространенность
ХЛЛ - одна из самых частых разновидностей лейкозов (30% всех случаев).

Этиология
• Вирусная инфекция (Эпштейн-Барра)
• Хромосомные аномалии в области 12, 13, 14-й
хромосом.
•
Этиология
• Вирусная инфекция (Эпштейн-Барра)
• Хромосомные аномалии в области 12, 13, 14-й
хромосом.
•

Патогенез
• отсутствуют признаки опухолевой прогрессии (бластный криз очень редко возникает в терминальной
Патогенез
• отсутствуют признаки опухолевой прогрессии (бластный криз очень редко возникает в терминальной

Классификация ХЛЛ (по K.R. Rai, 1975)
• 0 - абсолютный лимфоцитоз без видимого
Классификация ХЛЛ (по K.R. Rai, 1975)
• 0 - абсолютный лимфоцитоз без видимого

Классификация ХЛЛ (по J.L. Binet, 1977)
Стадия :
А(выживаемость > 10 лет)
Классификация ХЛЛ (по J.L. Binet, 1977)
Стадия :
А(выживаемость > 10 лет)

В зависимости от клинической картины
• опухолевый - периферические лимфатические узлы резко увеличены,
В зависимости от клинической картины
• опухолевый - периферические лимфатические узлы резко увеличены,

Клиника
• Лимфопролиферативный, обусловленный лимфаденопатией, спленомегалией и лимфоидной пролиферацией костного мозга:
- общие симптомы, обусловленные
Клиника
• Лимфопролиферативный, обусловленный лимфаденопатией, спленомегалией и лимфоидной пролиферацией костного мозга:
- общие симптомы, обусловленные

Клиника
• Синдром осложнений:
- гнойно-воспалительных;
- аутоиммунных (аутоиммунная гемолитическая анемия). Различная выраженность синдромов на тех или
Клиника
• Синдром осложнений:
- гнойно-воспалительных;
- аутоиммунных (аутоиммунная гемолитическая анемия). Различная выраженность синдромов на тех или

Диагностика
В общем анализе крови:
• количество лейкоцитов от 50х109/л до 100 х
Диагностика
В общем анализе крови:
• количество лейкоцитов от 50х109/л до 100 х
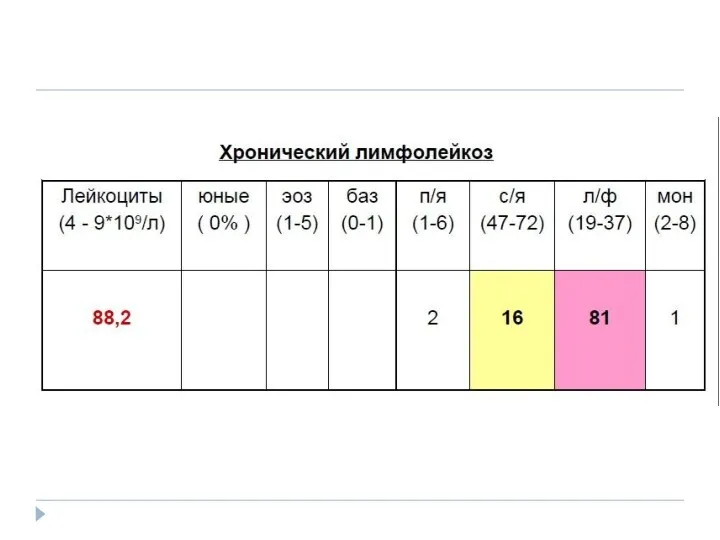

Примерно у 50% больных ХЛЛ обнаруживается
нормохромная нормоцитарная анемия.
Этиология:
• лейкозная инфильтрация костного
Примерно у 50% больных ХЛЛ обнаруживается
нормохромная нормоцитарная анемия.
Этиология:
• лейкозная инфильтрация костного

Диагностика
2) Биохимический анализ крови:
определение содержания белка, белковых фракций,
билирубина, аминотрансфераз, щелочной
фосфатазы, глюкоза,
Диагностика
2) Биохимический анализ крови:
определение содержания белка, белковых фракций,
билирубина, аминотрансфераз, щелочной
фосфатазы, глюкоза,

Диагностика
4) Иммунофенотипирование лимфоцитов!!!! –
ведущий метод диагностика – определение – CD-
кластеров на
Диагностика
4) Иммунофенотипирование лимфоцитов!!!! –
ведущий метод диагностика – определение – CD-
кластеров на

Стернальная пункция
Выраженная лимфоидная инфильтрация.
Лимфоциты составляют более 30% (иногда 50-60% и даже
Стернальная пункция
Выраженная лимфоидная инфильтрация.
Лимфоциты составляют более 30% (иногда 50-60% и даже

Лечение
• режим
• цитостатическая терапия
• лечебный лимфоцитоферез
• лучевая терапия
• спленэктомия
• ГКС
• лечение
Лечение
• режим
• цитостатическая терапия
• лечебный лимфоцитоферез
• лучевая терапия
• спленэктомия
• ГКС
• лечение

Показания к цитостатической терапии
1) Появление симптомов интоксикации;
2) Появление анемии или тромбоцитопении;
3)
Показания к цитостатической терапии
1) Появление симптомов интоксикации;
2) Появление анемии или тромбоцитопении;
3)

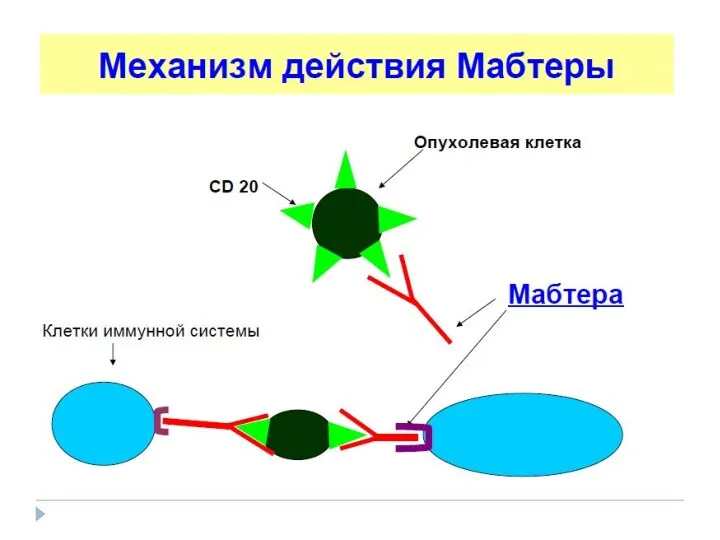
Современные подходы к терапии ХЛЛ
• Ритуксимаб (Мабтера) -
моноклональное антитело к антигену
СD
Современные подходы к терапии ХЛЛ
• Ритуксимаб (Мабтера) -
моноклональное антитело к антигену
СD
 Первая медицинская помощи при ранениях
Первая медицинская помощи при ранениях Мышцы нижней конечности
Мышцы нижней конечности Класифікація захворювань, що передаються статевим шляхом
Класифікація захворювань, що передаються статевим шляхом Гендік инженерия негіздері
Гендік инженерия негіздері Федеральный центр сердечно-сосудистой хирургии
Федеральный центр сердечно-сосудистой хирургии Бедеу жұпты тексеру алгоритмі
Бедеу жұпты тексеру алгоритмі Анестезиологическое обеспечение обширных ортопедических вмешательств
Анестезиологическое обеспечение обширных ортопедических вмешательств Абсцесс мягких тканей. Флегмона. Лимфаденит. Карбукул и фурункул у детей
Абсцесс мягких тканей. Флегмона. Лимфаденит. Карбукул и фурункул у детей Публичная декларация целей и задач Министерства здравоохранения Республики Коми на 2018 год
Публичная декларация целей и задач Министерства здравоохранения Республики Коми на 2018 год Пиодермии. Способствующие факторы
Пиодермии. Способствующие факторы Множественные аллели. Иммуногенетика (АВО, Rh, HLA). Формы взаимодействия генов. (Лекция 5)
Множественные аллели. Иммуногенетика (АВО, Rh, HLA). Формы взаимодействия генов. (Лекция 5) Балалар ауруларын біріктіріп жүргізу жоспарын құрастыру (жөтел және қиындаған тыныс)
Балалар ауруларын біріктіріп жүргізу жоспарын құрастыру (жөтел және қиындаған тыныс) The Heart
The Heart Зондовый массаж
Зондовый массаж Гель для выравнивания тона кожи Безупречная кожа
Гель для выравнивания тона кожи Безупречная кожа Медицинские критерии смерти человека
Медицинские критерии смерти человека Психологиялық көмек көрсету ерекшеліктері
Психологиялық көмек көрсету ерекшеліктері Остеопороз. Диагностика
Остеопороз. Диагностика Острая травма кровеносных сосудов
Острая травма кровеносных сосудов Гематуриялық синдром
Гематуриялық синдром Стероидные и нестероидные противовоспалительные средства
Стероидные и нестероидные противовоспалительные средства Психологія відчуття та сприйняття
Психологія відчуття та сприйняття Остеоартроз: просто о сложном
Остеоартроз: просто о сложном ДЦП: функциональная классификация, возможности реабилитации
ДЦП: функциональная классификация, возможности реабилитации Всемирный день борьбы со СПИДом
Всемирный день борьбы со СПИДом Саркоидоз легких
Саркоидоз легких Принципы рациональной антикоагулянтной терапии в травматологии, ортопедии и хирургии повреждений
Принципы рациональной антикоагулянтной терапии в травматологии, ортопедии и хирургии повреждений Явления шока
Явления шока