- Клиническая биохимия азотистого обмена

Содержание
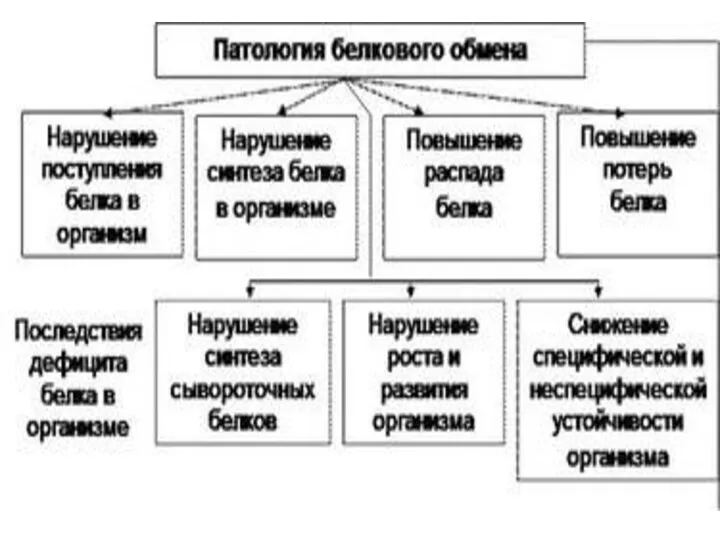
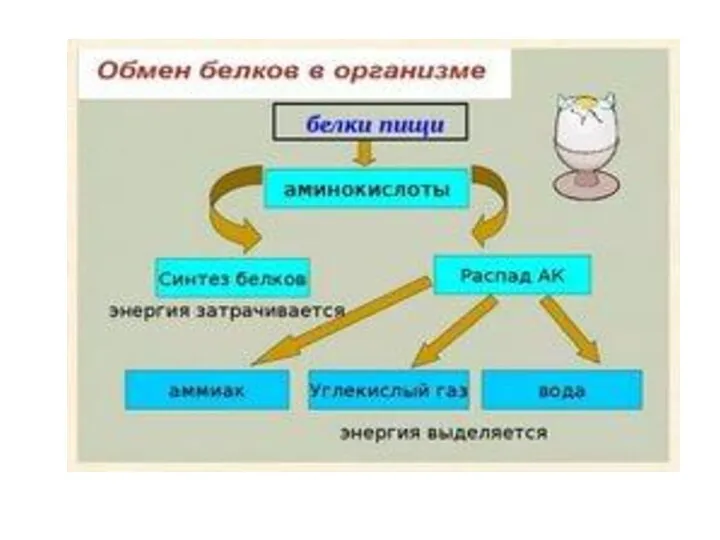
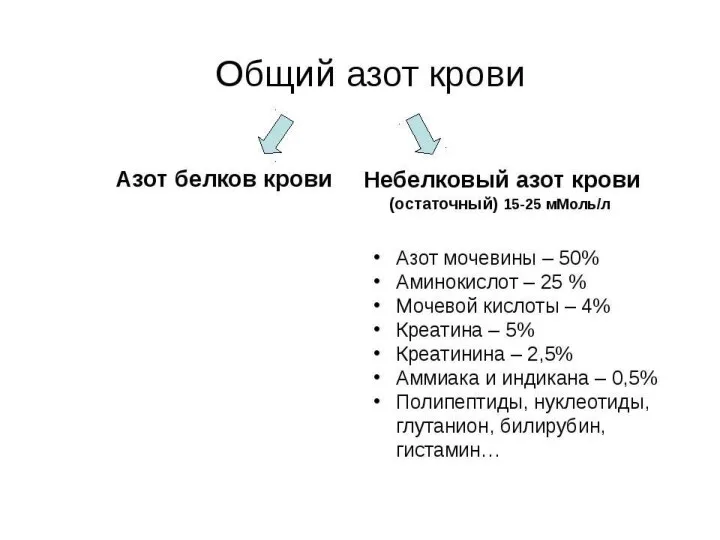
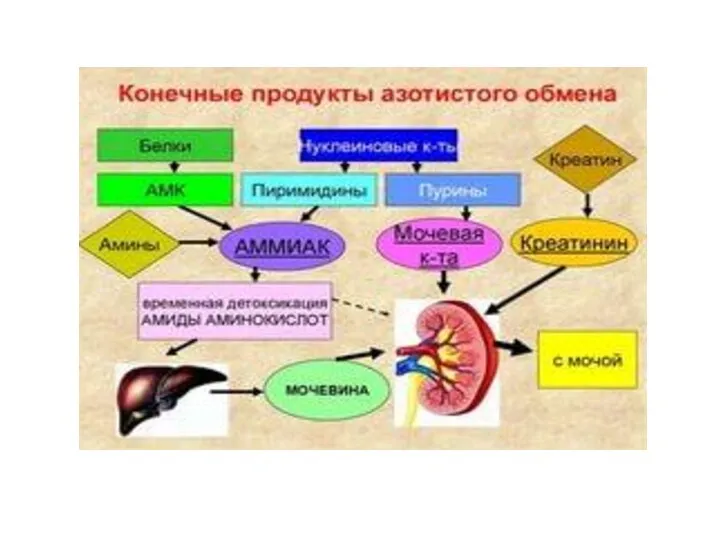
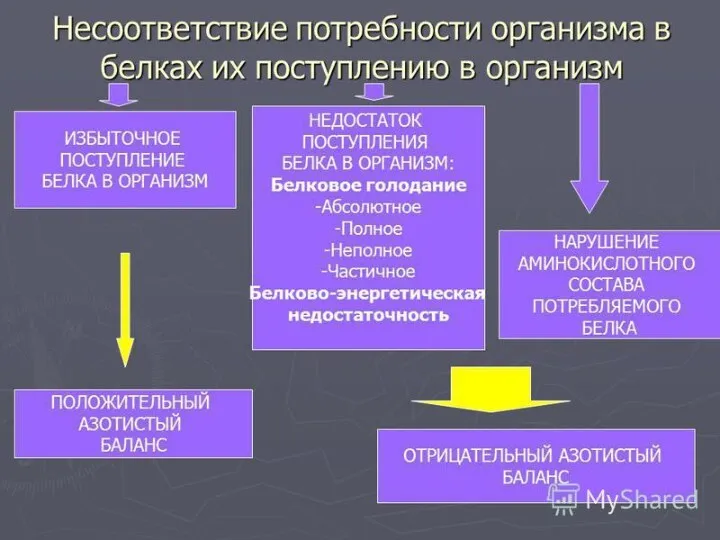
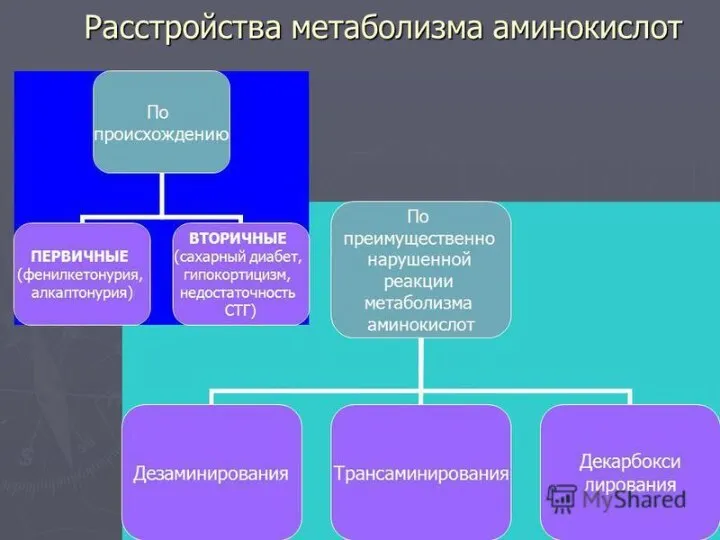
- 18. СПЕЦИФИЧЕСКИЕ ПУТИ ОБМЕНА АМИНОКИСЛОТ Эти пути обмена определяются различиями в строении радикалов аминокислот /АК/, поэтому они
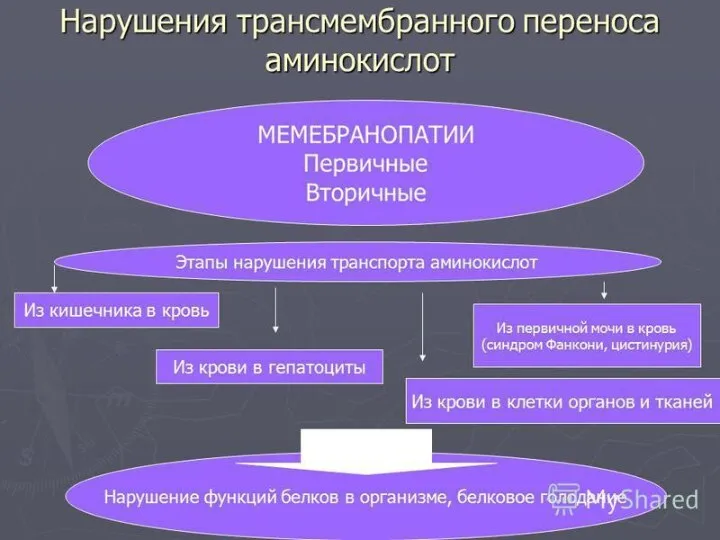
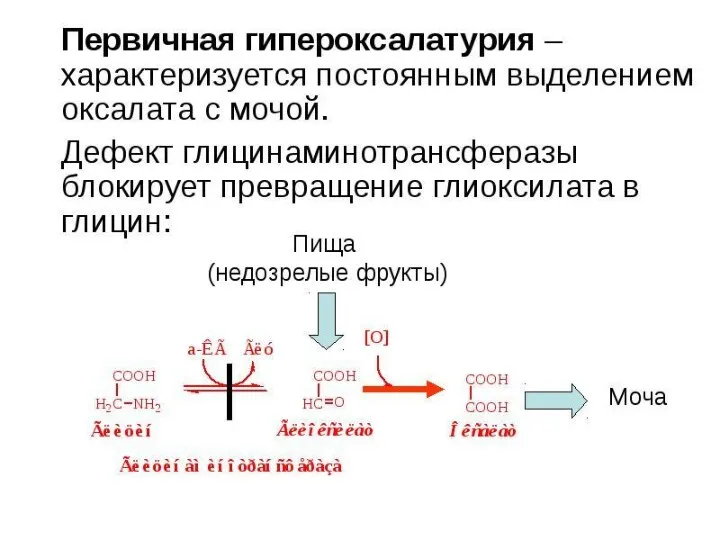
- 32. Наследственные дефекты всасывания АМК в почках Цистиноз (синдром Абдергальдена-Фанкони) Основной метаболический дефект - врожденное нарушение реабсорбции
- 33. Цистинурия (цистин-лизинурия) наследственное заболевание нарушение обратного всасывания цистина, лизина, аргинина и орнитина Экскреция с мочой цистина,
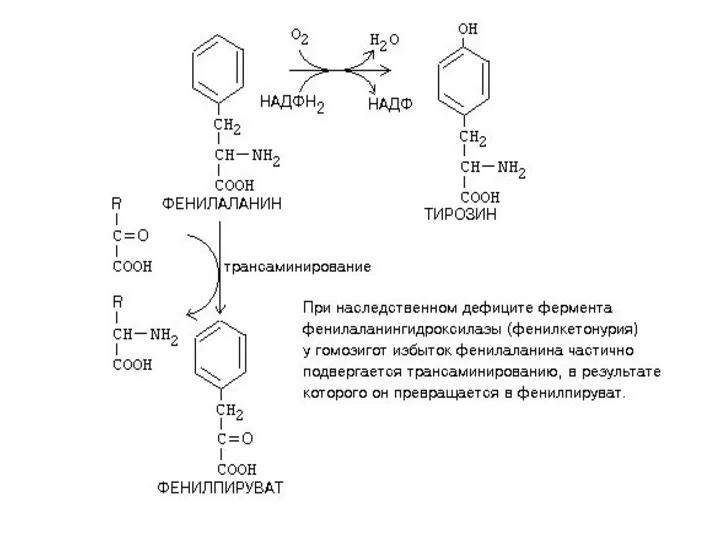
- 36. ОБМЕН ЦИКЛИЧЕСКИХ АМИНОКИСЛОТ ФЕНИЛАЛАНИНА И ТИРОЗИНА Фенилаланин - незаменимая аминокислота, тирозин - заменимая аминокислота. Фенилаланин вступает
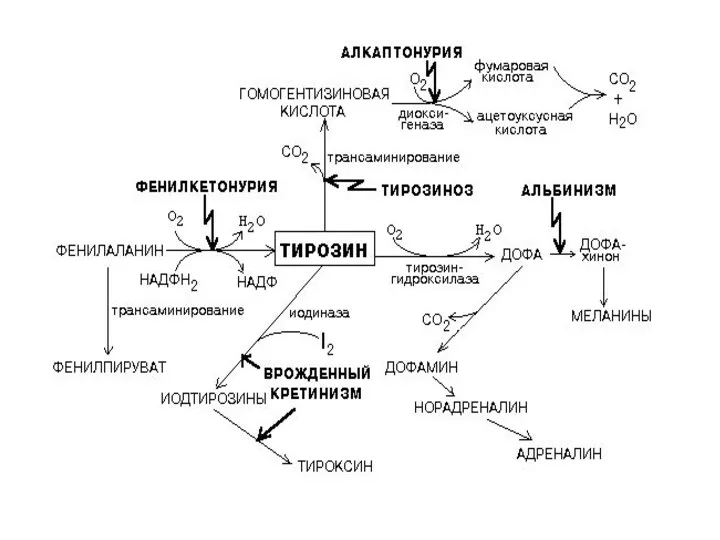
- 38. Из тирозина образуются: а) гормоны мозгового слоя надпочечников - адреналин и норадреналин, б) меланины - пигменты
- 40. НАРУШЕНИЯ ОБМЕНА ФЕНИЛАЛАНИНА И ТИРОЗИНА Нарушения обмена этих АК связано с нарушением биосинтеза некоторых ферментов, которые
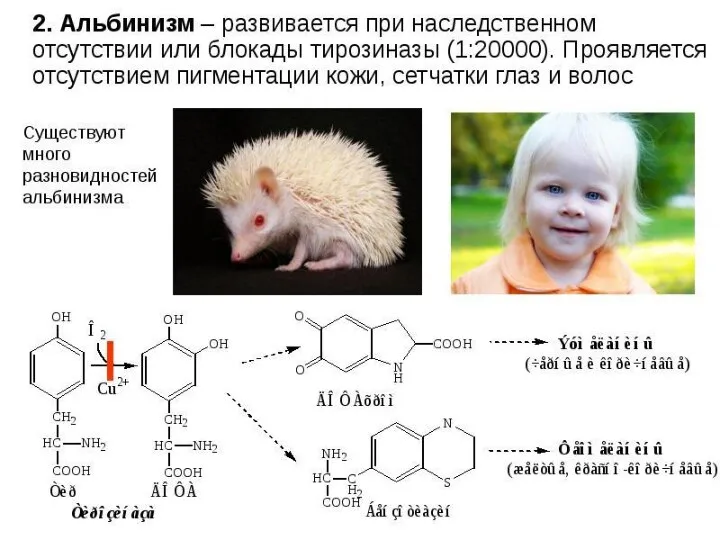
- 41. 2) альбинизм - нарушен синтез ферментов, превращающих ДОФА в ДОФА-хром, поэтому нарушается синтез меланинов. 3) алкаптонурия
- 42. Классификация ФКУ, обусловленной дефицитом ФАГ, в зависимости от уровня фенилаланина крови Форма заболевания Уровень фенилаланина в
- 43. Классическая фенилкетонурия (фенилкетонурия I типа) дефицит фермента фенилаланингидроксилазы (ФАГ) накопление фенилаланина и продуктов его распада в
- 44. Фенилкетонурия II типа дефицит дигидроптеридинредуктазы (DHPR) метаболические блоки - на путях превращения фенилаланина в тирозин, образования
- 45. Фенилкетонурия III типа Недостаточность 6-пирувоилтетрагидроптеринсинтазы (PTPS), участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптерин трифосфата Мутация структурного
- 49. Флюориметрия – количественный биохимический метод определения фенилаланина в крови методом хроматографии с помощью современных автоматических флюориметров.
- 50. Тандемная масс-спектрометрия – аналитический метод исследования, основанный на масс-спектрометрическом измерении. Проведение неонатального скрининга. Метод позволяет одновременно
- 52. Лечение фенилкетонурии: из рациона ребенка исключают фенилаланин и увеличивают в пище количество тирозина. Если ребенка держать
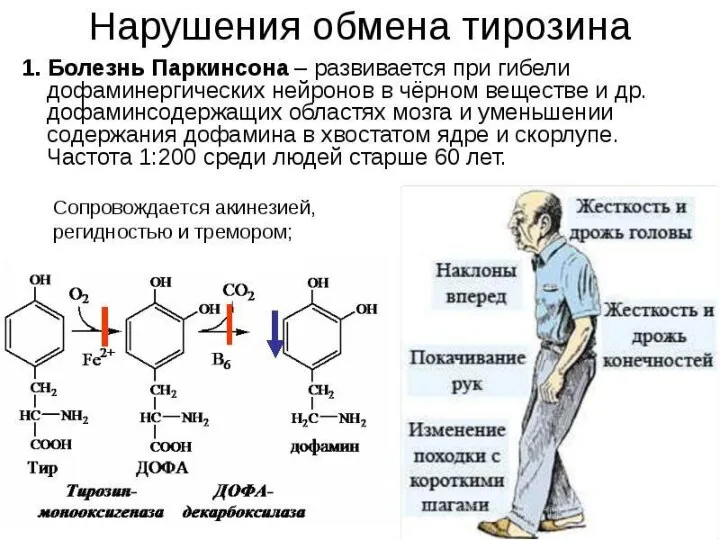
- 57. Наследственная тирозинемия 1 типа (НТ1) или гепаторенальная тирозинемия редкое (орфанное) генетическое заболевание с аутосомно-рецессивным типом наследования,
- 58. Врожденный дефект фермента фумарилацетоацетазы (фумарилацетогидролазы, FAH), осуществляющего в норме конечный этап деградации тирозина на нетоксичные фумарат
- 59. Патогенез НТ1 типа интоксикация продуктами аномального распада тирозина - фумарилацетоацетатом и малеилацетоацетатом и их конечными метаболитами
- 60. Сукцинилацетон ингибирует δ-аминолевулинат дегидратазу, (промежуточный медиатор порфобилиногена) нарушение биосинтеза гема клинически может проявляться симптомами острой перемежающей
- 61. Патогномоничный признак НТ1 - высокий уровень сукцинилацетона в моче и плазме крови (норма 0-2 мМоль/Моль креатинина).
- 62. В биохимических анализах крови умеренно повышенный уровень трансаминаз 2-3 нормы, признаки холестаза – высокий уровень ГГТП
- 63. Синдром Фанкони – Глюкозурия Фосфатурия Кальциурия Генерализованная аминоацидурия Почечный канальцевый ацидоз
- 64. 5. Исследование системы свертывания крови – витамин К – зависимая коагулопатия – дефицит всех печеночных факторов
- 65. Тирозинемия типа II недостаточность тирозин-аминотрансферазы (16q22.1-q22.3, ген ТАТ). Клинические проявления возникают в раннем возрасте: задержка умственного
- 66. Тирозинемия типа III недостаточность 4-гидроксифенилпируватгидроксилазы (12q24-qter, ген ЯРО). Характерны отставание в развитии, эпизоды атаксии, метаболический ацидоз.
- 72. Скачать презентацию











СПЕЦИФИЧЕСКИЕ ПУТИ ОБМЕНА АМИНОКИСЛОТ
Эти пути обмена определяются различиями в строении радикалов
СПЕЦИФИЧЕСКИЕ ПУТИ ОБМЕНА АМИНОКИСЛОТ
Эти пути обмена определяются различиями в строении радикалов












Наследственные дефекты всасывания АМК в почках
Цистиноз (синдром Абдергальдена-Фанкони)
Основной метаболический дефект -
Наследственные дефекты всасывания АМК в почках
Цистиноз (синдром Абдергальдена-Фанкони)
Основной метаболический дефект -

Цистинурия (цистин-лизинурия)
наследственное заболевание
нарушение обратного всасывания цистина, лизина, аргинина и орнитина
Экскреция
Цистинурия (цистин-лизинурия)
наследственное заболевание
нарушение обратного всасывания цистина, лизина, аргинина и орнитина
Экскреция


ОБМЕН ЦИКЛИЧЕСКИХ АМИНОКИСЛОТ
ФЕНИЛАЛАНИНА И ТИРОЗИНА
Фенилаланин - незаменимая аминокислота,
тирозин -
ОБМЕН ЦИКЛИЧЕСКИХ АМИНОКИСЛОТ
ФЕНИЛАЛАНИНА И ТИРОЗИНА
Фенилаланин - незаменимая аминокислота,
тирозин -

Из тирозина образуются:
а) гормоны мозгового слоя надпочечников -
адреналин и норадреналин,
б)
Из тирозина образуются:
а) гормоны мозгового слоя надпочечников -
адреналин и норадреналин,
б)

НАРУШЕНИЯ ОБМЕНА ФЕНИЛАЛАНИНА И ТИРОЗИНА
Нарушения обмена этих АК связано с нарушением
НАРУШЕНИЯ ОБМЕНА ФЕНИЛАЛАНИНА И ТИРОЗИНА
Нарушения обмена этих АК связано с нарушением

2) альбинизм - нарушен синтез ферментов, превращающих ДОФА в ДОФА-хром, поэтому
2) альбинизм - нарушен синтез ферментов, превращающих ДОФА в ДОФА-хром, поэтому

Классификация ФКУ, обусловленной дефицитом ФАГ,
в зависимости от уровня фенилаланина крови
Классификация ФКУ, обусловленной дефицитом ФАГ,
в зависимости от уровня фенилаланина крови

Классическая фенилкетонурия (фенилкетонурия I типа)
дефицит фермента фенилаланингидроксилазы (ФАГ)
накопление фенилаланина
Классическая фенилкетонурия (фенилкетонурия I типа)
дефицит фермента фенилаланингидроксилазы (ФАГ)
накопление фенилаланина

Фенилкетонурия II типа
дефицит дигидроптеридинредуктазы (DHPR)
метаболические блоки
- на путях превращения фенилаланина
Фенилкетонурия II типа
дефицит дигидроптеридинредуктазы (DHPR)
метаболические блоки
- на путях превращения фенилаланина

Фенилкетонурия III типа
Недостаточность 6-пирувоилтетрагидроптеринсинтазы (PTPS), участвующей в процессе синтеза тетрагидробиоптерина из
Фенилкетонурия III типа
Недостаточность 6-пирувоилтетрагидроптеринсинтазы (PTPS), участвующей в процессе синтеза тетрагидробиоптерина из




Флюориметрия –
количественный биохимический метод определения фенилаланина в крови методом хроматографии
с
Флюориметрия –
количественный биохимический метод определения фенилаланина в крови методом хроматографии
с

Тандемная масс-спектрометрия –
аналитический метод исследования, основанный на масс-спектрометрическом измерении.
Проведение
Тандемная масс-спектрометрия –
аналитический метод исследования, основанный на масс-спектрометрическом измерении.
Проведение


Лечение фенилкетонурии:
из рациона ребенка исключают фенилаланин и увеличивают в пище количество
Лечение фенилкетонурии:
из рациона ребенка исключают фенилаланин и увеличивают в пище количество



Наследственная тирозинемия 1 типа (НТ1) или гепаторенальная тирозинемия
редкое (орфанное) генетическое заболевание
Наследственная тирозинемия 1 типа (НТ1) или гепаторенальная тирозинемия
редкое (орфанное) генетическое заболевание

Врожденный дефект фермента фумарилацетоацетазы (фумарилацетогидролазы, FAH), осуществляющего в норме конечный этап
Врожденный дефект фермента фумарилацетоацетазы (фумарилацетогидролазы, FAH), осуществляющего в норме конечный этап

Патогенез НТ1 типа
интоксикация продуктами аномального распада тирозина - фумарилацетоацетатом и
Патогенез НТ1 типа
интоксикация продуктами аномального распада тирозина - фумарилацетоацетатом и

Сукцинилацетон
ингибирует δ-аминолевулинат дегидратазу, (промежуточный медиатор порфобилиногена)
нарушение биосинтеза гема
клинически может проявляться
Сукцинилацетон
ингибирует δ-аминолевулинат дегидратазу, (промежуточный медиатор порфобилиногена)
нарушение биосинтеза гема
клинически может проявляться

Патогномоничный признак НТ1 - высокий уровень сукцинилацетона в моче и плазме

В биохимических анализах крови
умеренно повышенный уровень трансаминаз 2-3 нормы,
умеренно повышенный уровень трансаминаз 2-3 нормы,

Синдром Фанкони –
Глюкозурия
Фосфатурия
Кальциурия
Генерализованная аминоацидурия
Почечный канальцевый ацидоз
Глюкозурия
Фосфатурия
Кальциурия
Генерализованная аминоацидурия
Почечный канальцевый ацидоз

5. Исследование системы свертывания крови –
витамин К – зависимая коагулопатия
витамин К – зависимая коагулопатия

Тирозинемия типа II
недостаточность тирозин-аминотрансферазы
(16q22.1-q22.3, ген ТАТ).
Клинические проявления возникают в
Тирозинемия типа II
недостаточность тирозин-аминотрансферазы
(16q22.1-q22.3, ген ТАТ).
Клинические проявления возникают в

Тирозинемия типа III
недостаточность 4-гидроксифенилпируватгидроксилазы
(12q24-qter, ген ЯРО).
Характерны отставание в
Тирозинемия типа III
недостаточность 4-гидроксифенилпируватгидроксилазы
(12q24-qter, ген ЯРО).
Характерны отставание в




 Полиомиелит. Протекание болезни полиомиелита
Полиомиелит. Протекание болезни полиомиелита Дисфункционалдық жатырдан қан кету
Дисфункционалдық жатырдан қан кету Интерпретация данных методов инструментальной диагностики ЭЭГ психических расстройств, связанных с органическим поражением ЦНС
Интерпретация данных методов инструментальной диагностики ЭЭГ психических расстройств, связанных с органическим поражением ЦНС Временная остановка наружного кровотечения. Ошибки на догоспитальном этапе
Временная остановка наружного кровотечения. Ошибки на догоспитальном этапе Акушерлік перитонит. Жайылған септикалық инфекция
Акушерлік перитонит. Жайылған септикалық инфекция Синдром хронической болезни почек
Синдром хронической болезни почек Балалар ауруларын біріктіріп жүргізу жоспарын құрастыру(жөтел және қиындаған тыныс)
Балалар ауруларын біріктіріп жүргізу жоспарын құрастыру(жөтел және қиындаған тыныс) Иммундық процестердің бұзылуы. Аллергия, анафилаксия, СПИД
Иммундық процестердің бұзылуы. Аллергия, анафилаксия, СПИД Сестринский процесс при гломерулонефритах
Сестринский процесс при гломерулонефритах Гинекологиялық науқастардан анамнез жинау
Гинекологиялық науқастардан анамнез жинау Роль ствола мозга в регуляции двигательных функций
Роль ствола мозга в регуляции двигательных функций Хронический гастрит
Хронический гастрит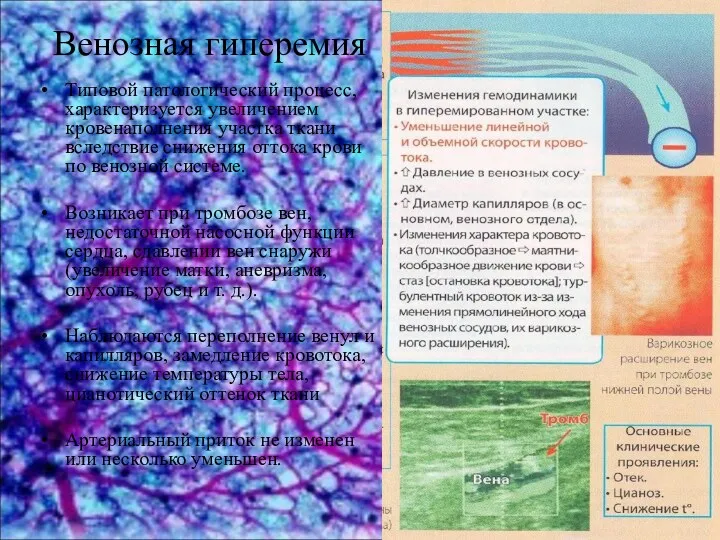 Венозная гиперемия
Венозная гиперемия Электронная медицинская аппаратура
Электронная медицинская аппаратура Лечение заболеваний и травм коленных суставов
Лечение заболеваний и травм коленных суставов Диагностические и профилактические мероприятия болезней вымени
Диагностические и профилактические мероприятия болезней вымени ЛФК при заболеваниях органов пищеварения
ЛФК при заболеваниях органов пищеварения Первая помощь при неотложных состояниях: закон и порядок
Первая помощь при неотложных состояниях: закон и порядок Нарушение кровообращения. Отеки
Нарушение кровообращения. Отеки Лайелл синдромы, Стивен-Джонсон синдромы
Лайелл синдромы, Стивен-Джонсон синдромы Профессиональные нейротоксикозы
Профессиональные нейротоксикозы Азық қорыту жүйесін зерттеу. Азық қабылдау және су ішудің бұзылуы
Азық қорыту жүйесін зерттеу. Азық қабылдау және су ішудің бұзылуы IgA нефропатия. Клиникасы:
IgA нефропатия. Клиникасы: Биохимия почек и мочи. (Лекция 10)
Биохимия почек и мочи. (Лекция 10) Укусы ядовитых животных
Укусы ядовитых животных Көз жасы мүшесінің патологиясы. Дакриоцистит, жас нүктесінің тарылуы, жас нүктесінің сырт айналуы
Көз жасы мүшесінің патологиясы. Дакриоцистит, жас нүктесінің тарылуы, жас нүктесінің сырт айналуы Парвавирусный энтерит собак
Парвавирусный энтерит собак Наследственные болезни обмена веществ
Наследственные болезни обмена веществ