- Клиническая генетика

Содержание
- 2. Основной целью медицинской генетики является изучение роли генетических составляющих в этиологии и патогенезе различных заболеваний человека
- 3. Эти болезни делятся на 3 класса: (1) наследственные болезни, (2) болезни с наследственной предрасположенностью, которые называют
- 4. Причиной развития наследственных болезней является присутствие в половых клетках родителей патологических мутаций, которые могут затрагивать хромосомы
- 5. Суммарная частота наследственных заболеваний достигает 1,5%, из них на долю хромосомных болезней приходится 0,5% и на
- 6. Отсутствие повторных случаев болезни у членов одной и той же семьи не исключает наследственного характера заболевания,
- 7. Врожденные заболевания могут быть как наследственными, так и приобретенными, например под действием тератогенных факторов или осложнений
- 8. Семейными называются болезни, присутствующие у нескольких членов одной семьи. Они могут быть наследственными или приобретенными, например,
- 9. В этиологии многофакторных заболеваний наряду с действием неблагоприятных внешних факторов существенное влияние оказывают состояния не одного,
- 10. Количество этих генов, формирующих наследственную предрасположенность к заболеванию, иногда исчисляется десятками или даже сотнями. К многофакторным
- 11. Основной этиологический механизм приобретенных заболеваний связан с неблагоприятными внешними воздействиями, такими как травмы или инфекции
- 12. Однако в последнем случае часто не удается полностью исключить влияния генетических факторов, определяющих дифференциальную чувствительность различных
- 13. Многие химические соединения и физические воздействия способны оказывать тератогенный эффект на плод в процессе беременности, то
- 14. В 2-3% случаев новорожденные имеют различные пороки развития. Не все ВПР могут быть диагностированы сразу после
- 15. ВПР нервной, мочеполовой, эндокринной систем, органов чувств и т.д. могут быть выявлены еще позже. Таким образом,
- 16. ВПР могут возникнуть под действием инфекционных агентов, таких как цитомегаловирус, краснуха, токсоплазмоз, вирус герпеса и др.,
- 17. Токсоплазменная эмбриофетопатия
- 18. Частота генитального хламидиоза у беременных женщин составляет 25%. Риск передачи инфекции ребенку равен 40-70%. Примерно 6-7%
- 19. Большую угрозу для здоровья будущего ребенка представляет краснуха. Если женщина перенесла это заболевание в первом триместре
- 20. Этот риск остается достаточно высоким в случае заболевания женщины во втором (25%) и в третьем (7-10%)
- 21. Краснушная эмбриофетопатия
- 22. Тератогенным эффектом обладают лекарственные препараты цитостатического и противосудорожного действия, стероидные гормоны, некоторые антибиотики, соли ртути, ретиноевая
- 23. Вальпроатная эмбриофетопатия
- 24. Повышена вероятность рождения детей с ВПР у матерей, страдающих аутоиммунными болезнями, сахарным диабетом, эпилепсией, гипотиреозом и
- 25. Диабетическая эмбриофетопатия
- 26. Тератогенным эффектом обладают большие дозы никотина и алкоголя, а также некоторые наркотические вещества (героин, кокаин и
- 27. Алкогольная эмбриофетопатия
- 28. Эффект тератогенов существенно зависит от стадии эмбриогенеза, то есть срока беременности, на котором плод подвергается такому
- 29. Первые две недели беременности являются критическим периодом для внутриутробного развития человека. При неблагоприятном воздействии в этот
- 30. Летальные бластопатии (циклопия, сиреномелия)
- 31. Следующий период активного органогенеза до 12 недели беременности является особенно чувствительным к действию тератогенов, которые могут
- 32. К эмбриопатиям относятся такие тяжелые ВПР как дефекты заращения нервной трубки (ДЗНТ) – анэнцефалия, черепномозговая грыжа
- 33. Дефекты заращения нервной трубки (анэнцефалия, спинномозговая грыжа, энцефалоцеле)
- 34. Различные варианты расщелины губы и неба
- 35. Опасность возникновения ВПР под действием тератогенов сохраняется и на следующих стадиях беременности, хотя тяжесть и частота
- 36. Примеры фетопатии (гидроцефалия, синдром амниотических перетяжек )
- 37. Причиной развития наследственных болезней является присутствие в половых клетках родителей патологических мутаций
- 38. Мутации могут быть геномными, хромосомными и генными. Числовые хромосомные мутации затрагивают целые хромосомы. К ним относятся
- 39. Основная масса зародышей (до 60%) с дисбалансом хромосом погибает в ранний период развития плода. У половины
- 40. У 5% детей, погибших в перинатальном периоде также обнаруживаются хромосомные аномалии. В 75% случаев – трисомии,
- 41. В 34% случаев у детей с хромосомными нарушениями обнаруживаются анеуплоидии по половым хромосомам, в 30% -
- 42. Трисомии среди живорожденных описаны лишь для шести хромосом, по остальным хромосомам они летальны. Из них наиболее
- 43. Лицевые аномалии при синдроме Дауна
- 44. Больные синдром Патау
- 45. Часто числовые аномалии затрагивают половые хромосомы. Присутствие дополнительной Х-хромосомы у мужчин приводит к синдрому Клайнфельтера, а
- 46. Синдром Клайнфельтера
- 47. Синдром Шерешевского-Тернера
- 48. Моногенные болезни обусловлены присутствием мутаций в одном гене. Следствием мутаций может быть нарушение структуры или синтеза
- 49. Число моногенных заболеваний достигает 5000. Наиболее частыми из них (1:2-3 до 1:10-20 тысяч) являются муковисцидоз, фенилкетонурия,
- 50. Среди моногенных болезней значительный процент составляют ферментопатии, различные формы умственной отсталости, дефекты органов слуха, зрения, скелетные
- 51. Моногенные заболевания в редких случаях встречаются среди таких нозологических форм, которые в общем случае не являются
- 52. Моногенные варианты заболевания, как правило, отличаются от спорадических форм более тяжелым течением и ранним дебютом
- 53. Несмотря на клиническое многообразие моногенных болезней, можно выделить некоторые общие черты, касающиеся возраста начала заболевания, характера
- 54. Большинство моногенных болезней распознаются в перинатальном или раннем детском возрасте. Около 25% этих болезней развиваются в
- 55. Некоторые моногенные болезни, такие как спиноцеребеллярные атаксии, миодистрофия Ландузи-Дежерина, хорея Гентингтона, моногеннные формы болезни Альцгеймера и
- 56. Типичными чертами многих наследственных заболеваний являются хронический характер и прогредиентность течения
- 57. При некоторых моногенных заболеваниях выявляются редкие специфические симптомы, проявления которых не имеют клинического значения, но являются
- 58. Внешний вид больных часто столь специфичен, что делает их более похожими друг на друга, чем на
- 59. Мукополисахаридоз I типа
- 60. При синдроме Вильямса необычное лицо «эльфа» создается коротким носом, эпикантом, длинным фильтром и полными щеками
- 61. Синдром Вильямса
- 62. Черепно-лицевые особенности при синдроме Рассела-Сильвера
- 63. Моногенные заболевания классифицируют по типам наследования, которые в большинстве случаев соответствуют законам Менделя
- 64. Наследование моногенных заболеваний зависит от характера доминирования и нахождения гена в аутосоме или в половой хромосоме.
- 65. Аутосомно-доминантный тип наследования
- 66. Особенности аутосомно-доминантного наследования Болеют в равной степени мужчины и женщины Как правило, больные являются гетерозиготными носителями
- 67. Около 50% врожденных пороков развития относятся к аутосомно-доминантным заболеваниям. По аутосомно-доминантному типу наследуются синдром Марфана, большинство
- 68. Однако самой многочисленной группой аутосомно-доминантных заболеваний являются наследственные опухолевые синдромы. Их суммарная частота в популяциях составляет
- 69. Единственным клиническим проявлением наследственных опухолевых синдромов является повышенная вероятность возникновения онкологических заболеваний, которая с возрастом может
- 70. Наиболее известным аутосомно-доминантным заболеванием является синдром Марфана, при котором у больных наблюдается одновременное поражение трех систем:
- 71. Характерными клиническими проявлениями синдрома Марфана являются высокий рост, в сочетании с выраженным сколиозом или лордозом, арахнодактилия,
- 73. Предполагали, что заболевание обусловлено мутациями в одном из коллагеновых генов. Однако оказалось, что при синдроме Марфана
- 74. Факоматозы характеризуются сочетанным поражением нервной системы, кожных покровов и внутренних органов. Среди них самым известным является
- 75. Характерными клиническими проявлениями нейрофиброматоза I являются доброкачественные опухоли кожи и подкожной клетчатки – нейрофибромы. Часто наблюдаются
- 77. В группу наследственных коллагенопатий, обусловленных мутациями в генах коллагенов и ферментов их биосинтеза, входят более 70
- 78. Коллагены составляют более 30% общей массы белков тела млекопитающих. Разнообразие коллагеновых белков достаточно велико (всего 27
- 79. Все коллагеновые альфа-цепи имеют коллагеновый домен, на протяжении которого каждая третья аминокислота является глицином. Такое расположение
- 81. Более 90% коллагеновых волокон образованы мажорными фибриллярными коллагенами I, II и III типов
- 82. Коллаген I типа экспрессируется повсеместно, но особенно обильно представлен в костной системе, сухожилиях и коже. Гетерозиготные
- 83. Клиническая картина несовершенного остеогенеза характеризуется повышенной ломкостью костей и патологическими изменениями ряда других тканей, богатых коллагеном
- 84. Наблюдается высокий клинический полиморфизм заболевания от летальных неонатальных до взрослых форм, при которых множественные переломы костей
- 85. Неонатальная форма несовершенного остеогенеза
- 86. Девочка 18 лет с тяжелой формойа несовершенного остеогенеза
- 87. Оказалось, что при тяжелых формах заболевания часто обнаруживаются миссенс-мутации, изменяющие положения глицина. При этом происходит неправильное
- 88. При легких формах частыми являются нонсенс-мутации. При этом мутантная альфа-цепь деградирует и не участвует в формировании
- 89. Коллаген II типа является мажорным хрящевым коллагеном и составляет основу стекловидного тела. Гетерозиготные мутации в гене
- 90. Спондилоепиметафизарная дисплазия
- 91. Спондилоепиметафизарная дисплазия (два брата)
- 92. Синдром Элерса-Данло характеризуется гиперрастяжимостью и истончением кожи, гипермобильностью суставов, скелетными аномалиями, неровным ростом зубов, деформацией ногтей
- 93. Клинические проявления синдрома Элерса-Данло
- 94. Скелетные аномалии при синдроме Элерса-Данло
- 95. Семейный случай синдрома Элерса-Данло
- 96. Описано около 10 наследственных вариантов синдрома Элерса-Данло. Классические формы заболевания обусловлены дефектами коллагена V типа.
- 97. Самая тяжелая «артериальная» форма синдрома Элерса-Данло, которая может сопровождаться разрывами артерий и перфорацией внутренних органов, обусловлена
- 98. Аутосомно-рецессивный тип наследования
- 99. Особенности аутосомно-рецессивного наследования Больные дети являются гомозиготными носителями мутаций Они рождаются с вероятностью 25% у здоровых
- 100. Частоты гетерозигот по рецессивным заболеваниям с распространенностью: 1 на 2-20 000 –1:20-70, 1 на 30-100 000
- 101. Поэтому выдвигавшиеся в начале XX века евгенические предложения по стерилизации больных с рецессивной патологией с современных
- 102. По аутосомно-рецессивному типу наследуются болезни обмена (НБО) – одна из наиболее многочисленных и хорошо изученных групп
- 103. НБО обусловлены нарушением каталитической функции различных ферментов, участвующих в обмене аминокислот, углеводов, липидов, гликозаминогликанов, гормонов, пуринов
- 104. Подобные нарушения часто сопровождаются накоплением веществ, предшествующих ферментативному блоку, и дефицитом конечных продуктов реакции
- 105. Общими нарушениями при наследственных дефектах обмена аминокислот являются аминоацидурия (выделение аминокислот с мочей) и ацидоз тканей.
- 107. Гиперфенилаланинемии (ГФА) – это группа генетически гетерогенных аутосомно-рецессивных заболеваний, обусловленных нарушением метаболизма фенилаланина
- 108. В основе патогенеза ГФА лежит накопление в крови фенилаланина (незаменимой аминокислоты, которая не синтезируется в организме,
- 109. Фенилкетонурия (ФКУ), наиболее частая и злокачественная форма ГФА. Заболевание обусловлено наследственной недостаточностью фенилаланингидроксилазы. Частота ФКУ составляет
- 110. Ведущим симптомом болезни является отставание умственного развития – олигофрения. Уже с первых недель жизни ребенка наблюдаются
- 111. Лечение больных заключается в исключении из питания фенилаланина путем применения специфической безфенилаланиновой диеты
- 112. Успех лечения зависит от того, насколько рано поставлен диагноз. Поэтому всем новорожденным проводится обязательное централизованное скринирующее
- 113. Больные фенилкетонурией, выявленные по неонатальному скринигу
- 114. В России имеется мажорная миссенс-мутация R408W в гене фенилаланингидроксилазы (PAH), частота которой у больных достигает 60%,
- 115. Пренатальная диагностика мутации R408W в гене PAH методом рестрикционного анализа
- 116. Наиболее распространенным аутосомно-рецессивным заболеванием среди европейцев является муковисцидоз. Его частота в России составляет 1 на 3-5
- 117. Молекулярной основой патогенеза муковисцидоза является нарушение работы хлорного канала, локализованного на мембранах эпителиальных клеток
- 118. Самой распространенной мутаций в гене муковисцидоза (CFTR) является delF508 – делеция трех нуклеотидов в 10-ом экзоне
- 119. Присутствие мутации delF508 у больных и гетерозиготных носителей легко определить методом ПЦР и электрофореза, что позволяет
- 120. Пренатальная диагностика делеции delF508 в гене муковисцидоза (CFTR)
- 121. Спинальная мышечная атрофия I
- 122. Динамика почерка больной ГЛД на фоне лечения в течение 36 лет
- 123. Х-сцепленное рецессивное наследование
- 124. Х-сцепленное рецессивное наследование
- 125. Особенности Х-сцепленного рецессивного наследования Болеют только мальчики Оба родителя здоровы, но мать несет гетерозиготную мутацию в
- 126. Наиболее известными Х-сцепленными рецессивными заболеваниями являются гемофилия А и В, миодистрофия Дюшенна, синдром Мартина Белл и
- 127. Синдром Мартина Белл
- 129. Основным продуктом гена миодистрофии Дюшенна (DMD) является стержневидный белок дистрофин, располагающейся на цитоплазматической поверхности мембраны мышечного
- 130. Дистрофин – это полифункциональный белок, обеспечивающий поддержание целостности мембраны мышечного волокна при раундах сокращения-расслабления, а также
- 131. Структура дистрофина
- 132. В большинстве случаев у больных диагностируются протяженные внутригенные делеции в гене DMD, затрагивающие несколько соседних экзонов.
- 133. Диагностика делеций в гене DMD осуществляется методом мультиплексной ПЦР, при которой одновременно амплифицируются несколько внутригенных фрагментов
- 134. Пренатальная диагностика миодистрофии Дюшенна
- 135. Некоторые моногенные заболевания не подчиняется законам Менделя. Это митохондриальные заболевания, болезни экспансии и болезни, обусловленные нарушениями
- 136. Митохондриальный тип наследования
- 137. К Мт-болезням относятся синдром Лебера (атрофия зрительного нерва), MELAS-синдром (лактоацидоз с инсульт-подобными эпизодами), MERF-синдром (миоклонус-эпилепсия с
- 138. Многофакторные или комплексные заболевания обусловлены комбинированным действием неблагоприятных средовых и генетических факторов риска, формирующих наследственную предрасположенность
- 139. К ним относятся социально-значимыме хронические болезни, такие как сердечно-сосудистая патология, диабет, бронхиальная астма, внутренние болезни, психические,
- 140. В качестве генетических факторов риска многофакторной патологии рассматривают широко распространенные полиморфные аллели, обладающие относительно небольшим повреждающим
- 141. Поиск генов-кандидатов, формирующих «генную сеть» многофакторного заболевания, осуществляют, исходя из знаний об его этиологии и патогенезе
- 142. Что мы знаем о заболевании? Какие метаболические циклы дефектны при тех или иных заболеваниях? Какие белки
- 143. Есть ли там широко распространенные среди населения (полиморфные) аллели, влияющие на функцию гена, прежде всего, снижающие
- 144. Для того чтобы определить, является ли полиморфный аллель генетическим фактором риска, предрасполагающим к развитию определенного заболевания
- 145. Только в тех случаях, когда уровни полиморфизма среди больных оказываются достоверно выше по сравнению с контролем,
- 146. Идентификация генетических факторов риска и разработка индивидуальных профилактических мероприятий составляют стратегическую основу нового направления предиктивной медицины
- 147. Фармакогенетика – это раздел медицинской генетики, изучающий влияние наследственной конституции на метаболизм различных лекарственных препаратов с
- 148. По разным оценкам вклад генетических составляющих в вариабельность реакции на лекарственные препараты колеблется в пределах от
- 149. В настоящее время в рамках некоторых международных проектов (PharmacoGenetics for Every Nation Initiative) создаются базы данных
- 150. Описан ряд наследственных болезней обмена, ведущих к медикаментозным идиосинкразиям
- 151. Мутации в гене псевдохолинэстеразы препятствуют гидролизу используемого в анестезиологии препарата суксаметония. У гомозигот по редким аллелям,
- 152. В гене Г-6-ФДГ идентифицированы мутации, ассоциированные с различными формами гемолитической анемии. Описаны полиморфные аллели, при которых
- 153. Прием барбитуратов, сульфаниламидов, некоторых противосудорожных препаратов и антибиотиков может привести к развитию острой перемеживающейся порфирии у
- 154. Ее клиническими проявлениями являются острые боли в животе, красный цвет мочи, анурия, периферические невриты и параличи,
- 155. Наследуется заболевание по аутосомно-доминантному типу с неполной пенетрантностью, его частота в некоторых популяциях достигает 1:10000
- 156. При некоторых наследственных заболеваниях может наблюдаться необычная реакция на определенные лекарственные препараты. Типичным примером является подагра,
- 157. Заболевание обусловлено ускоренным синтезом мочевой кислоты с одновременным снижением ее выведения почками. При этом в различных
- 158. Некоторые диуретики (хлортиазид, фуросемид) могут ускорить и резко усилить клинические проявления заболевания вплоть до развития гиперурикемии
- 159. Все рассмотренные выше примеры касались достаточно редких моногенных болезней. Однако различная индивидуальная реакция на лекарственные препараты,
- 160. В большинстве случаев она связана не с наследственными заболеваниями, а с присутствием полиморфных аллелей в генах
- 161. Одним из наиболее ярких примеров подобного рода является влияние скорости ацетилирования, осуществляемого ферментом N-ацетилтрансферазой 2, на
- 162. В гене NAT2 имеются полиморфные аллели, влияющие на активность соответствующего фермента. В зависимости от присутствия этих
- 163. В европейских популяциях соотношение между этими двумя группами населения примерно одинаково, тогда как среди желтой расы
- 164. «Медленные» ацетиляторы в большей степени склонны к интоксикации. В частности, при регулярном приеме противотуберкулезного препарата изониазида
- 165. Его накопление сопровождается дефицитом нейротропных витаминов группы В. Поэтому при терапии изониазидом обязательно необходимо назначать больным
- 166. «Медленные» ацетиляторы в большей степени склонны к развитию гемолитической анемии при приеме сульфаниламидов, а также некоторых
- 167. Дозы изониазида могут быть снижены, если больные относятся к группе «медленных» ацетиляторов. Среди «быстрых» ацетиляторов чаще
- 168. Ключевая роль в метаболизме многих лекарственных препаратов принадлежит цитохромам P450. Их разнообразие очень велико
- 169. Разберем более подробно связь цитохромов P450 с лекарственным метаболизмом на примере главного лекарственного метаболизатора – полипептида
- 170. Этот цитохром непосредственно взаимодействует с S-варфарином, который широко используется при лечении пациентов с тромботическими заболеваниями или
- 171. Успех лечения зависит от выбора дозы препарата и длительности курса. С учетом специфики заболевания и индивидуальных
- 172. В гене CYP2C9 идентифицированы два функциональных полиморфизма, влияющие на метаболическую активность этого цитохрома. Их частоты в
- 173. В отечественных популяциях суммарная частота носителей минорных аллелей гена CYP2C9 достигает 34%. В гомозиготном состоянии остаточная
- 174. Больные с низкой активностью метаболизатора имеют большую вероятность геморрагических осложнений при проведении варфариновой терапии. Для них
- 175. Значимыми с фармакологической точки зрения являются некоторые полиморфные аллели в других генах цитохромов, в частности в
- 176. Кроме того, в биотрансформации лекарственных препаратов принимают участие многие другие ферменты – глутатионтрансферазы, N-ацетилтрансферазы, моноаминооксидазы, холинэстеразы,
- 177. Одной из ведущих проблем современной фармакогенетики является разработка схем терапии различных заболеваний с учетом генотипического статуса
- 178. Описание различных этнических групп по специфическим профилям полиморфизмов генов лекарственного метаболизма может способствовать разработке национальных программ
- 179. Лечение больных с врожденной и наследственной патологией в основном носит симптоматический характер. Не влияя на причину
- 180. При этом могут быть использованы самые разнообразные методы терапии – медикаментозные, хирургические, физиотерапевтические и др.
- 181. Результаты симптоматического лечения, обычно, не очень продолжительны, требуется многократное повторение лечебных процедур, причем их эффективность со
- 182. Необходимыми элементами воспитания больных синдромом Дауна являются длительные упражнения по развитию мелкой моторики рук, регулярные занятия
- 183. Наряду с этим, желательно применение препаратов нооторопного действия, фолатов, витаминотераприя и т.д. В этих случаях можно
- 184. При синдроме Шерешевского-Тернера применяются гормональная терапия для стимуляции роста и препараты ноотропного действия. При мозаичном варианте
- 185. При наличии гинекомастии у мужчин с синдромом Клайнфельтера производят удаление молочных желез, а для преодоления бесплодия
- 186. Патогенетическое лечение наследственных болезней направлено на коррекцию биохимических и физиологических процессов, нарушение работы которых связано с
- 187. Самым простым методом патогенетического лечения является диетотерапия. Но она эффективна лишь в тех случаях, когда патологический
- 188. Наиболее эффективным способом терапевтической коррекции наследственных энзимопатий является введение недостающего фермента в те клетки больного, которые
- 189. Основными проблемами при этом являются: (1) получение в достаточном количестве чистого фермента, (2) преодоление реакции иммунологического
- 190. Большого успеха удалось достичь при лечении с помощью заместительной ферментотерапии болезни Гоше. Своевременно начатое лечение и
- 191. Хорошие результаты получены при заместительной ферментотерапии мукополисахаридозов, особенно в тех случаях, когда они не сопровождаются тяжелыми
- 192. При заболеваниях, обусловленных дефицитом конечного продукта патологической метаболической цепи, лечение направлено на возмещение недостающего продукта. Такой
- 193. Примером является лечение врожденного гипотиреоза тироксином, своевременное назначение которого полностью предотвращает развитие клинических симптомов
- 194. Наиболее перспективным способом этиологического лечения наследственных заболеваний является исправление генетических дефектов, то есть генотерапия
- 195. Количество генотерапевтических проектов, находящихся на стадии клинических испытаний, в США уже превышает 1500. В ряде центров
- 196. Объекты клинических испытаний проектов генной терапии
- 197. Программы генной терапии оказались наиболее успешны при лечении некоторых наследственных иммунодефицитов
- 198. Однако в настоящее время нет ни одного генотерапевтического проекта, который имел бы реальное клиническое значение для
- 199. Таким образом, генотерапия остается одним из наиболее привлекательных и перспективных направлений молекулярной медицины будущего
- 201. Скачать презентацию

Основной целью
медицинской генетики является изучение роли генетических составляющих
в этиологии
Основной целью медицинской генетики является изучение роли генетических составляющих в этиологии

Эти болезни делятся на 3 класса:
(1) наследственные болезни, (2) болезни
Эти болезни делятся на 3 класса: (1) наследственные болезни, (2) болезни

Причиной развития наследственных болезней является присутствие в половых клетках родителей патологических
Причиной развития наследственных болезней является присутствие в половых клетках родителей патологических

Суммарная частота наследственных заболеваний достигает 1,5%, из них на долю хромосомных
Суммарная частота наследственных заболеваний достигает 1,5%, из них на долю хромосомных

Отсутствие повторных случаев болезни у членов одной и той же семьи
Отсутствие повторных случаев болезни у членов одной и той же семьи

Врожденные заболевания могут быть как наследственными, так и приобретенными, например под
Врожденные заболевания могут быть как наследственными, так и приобретенными, например под

Семейными называются болезни, присутствующие у нескольких членов одной семьи. Они могут
Семейными называются болезни, присутствующие у нескольких членов одной семьи. Они могут

В этиологии многофакторных заболеваний наряду с действием неблагоприятных внешних факторов существенное
В этиологии многофакторных заболеваний наряду с действием неблагоприятных внешних факторов существенное

Количество этих генов, формирующих наследственную предрасположенность к заболеванию, иногда исчисляется десятками
Количество этих генов, формирующих наследственную предрасположенность к заболеванию, иногда исчисляется десятками

Основной этиологический механизм приобретенных заболеваний связан с неблагоприятными внешними воздействиями, такими
Основной этиологический механизм приобретенных заболеваний связан с неблагоприятными внешними воздействиями, такими

Однако в последнем случае часто не удается полностью исключить влияния генетических
Однако в последнем случае часто не удается полностью исключить влияния генетических

Многие химические соединения и физические воздействия способны оказывать тератогенный эффект на
Многие химические соединения и физические воздействия способны оказывать тератогенный эффект на

В 2-3% случаев новорожденные имеют различные пороки развития. Не все ВПР
В 2-3% случаев новорожденные имеют различные пороки развития. Не все ВПР

ВПР нервной, мочеполовой, эндокринной систем, органов чувств и т.д. могут быть
ВПР нервной, мочеполовой, эндокринной систем, органов чувств и т.д. могут быть

ВПР могут возникнуть под действием инфекционных агентов, таких как цитомегаловирус, краснуха,
ВПР могут возникнуть под действием инфекционных агентов, таких как цитомегаловирус, краснуха,
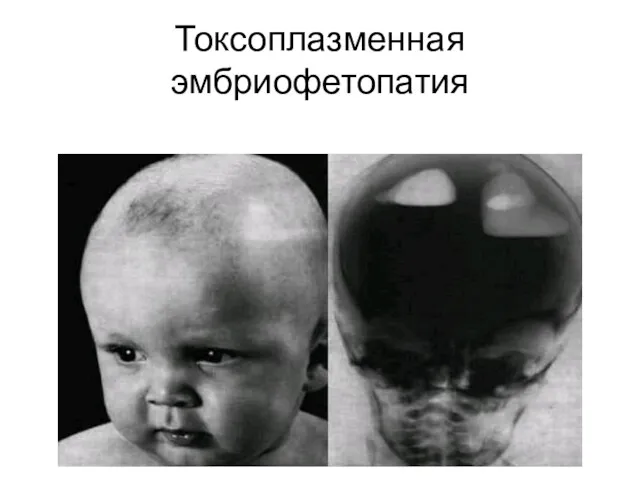
Токсоплазменная эмбриофетопатия
Токсоплазменная эмбриофетопатия

Частота генитального хламидиоза у беременных женщин составляет 25%. Риск передачи инфекции
Частота генитального хламидиоза у беременных женщин составляет 25%. Риск передачи инфекции

Большую угрозу для здоровья будущего ребенка представляет краснуха.
Если женщина перенесла
Большую угрозу для здоровья будущего ребенка представляет краснуха. Если женщина перенесла

Этот риск остается достаточно высоким в случае заболевания женщины во втором
Этот риск остается достаточно высоким в случае заболевания женщины во втором

Краснушная эмбриофетопатия
Краснушная эмбриофетопатия

Тератогенным эффектом обладают лекарственные препараты цитостатического и противосудорожного действия, стероидные гормоны,
Тератогенным эффектом обладают лекарственные препараты цитостатического и противосудорожного действия, стероидные гормоны,

Вальпроатная эмбриофетопатия
Вальпроатная эмбриофетопатия

Повышена вероятность рождения детей с ВПР у матерей, страдающих аутоиммунными болезнями,
Повышена вероятность рождения детей с ВПР у матерей, страдающих аутоиммунными болезнями,
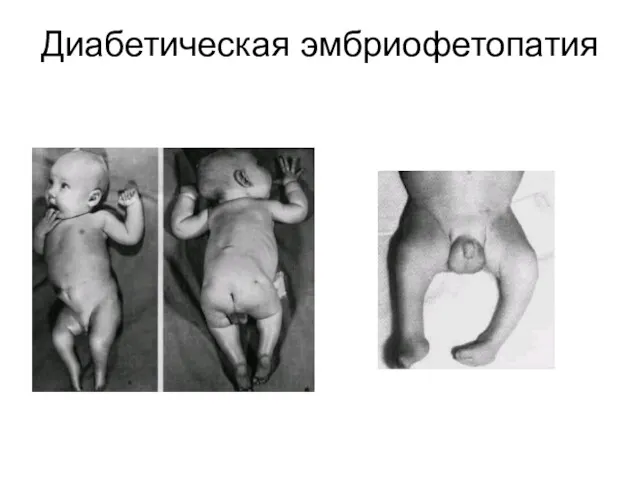
Диабетическая эмбриофетопатия
Диабетическая эмбриофетопатия

Тератогенным эффектом обладают большие дозы никотина и алкоголя, а также некоторые
Тератогенным эффектом обладают большие дозы никотина и алкоголя, а также некоторые
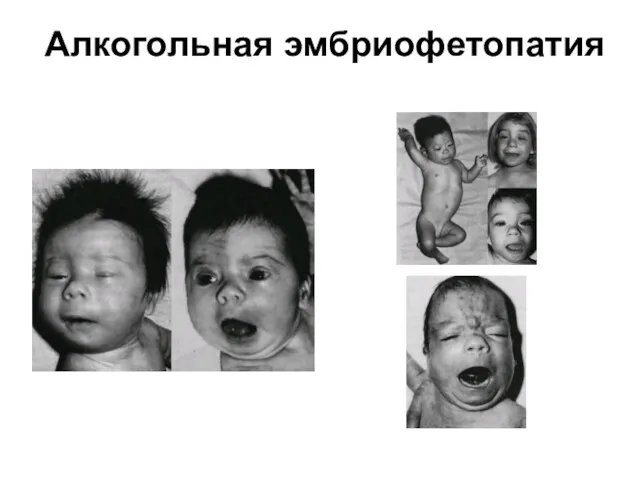
Алкогольная эмбриофетопатия
Алкогольная эмбриофетопатия

Эффект тератогенов существенно зависит от стадии эмбриогенеза, то есть срока беременности,
Эффект тератогенов существенно зависит от стадии эмбриогенеза, то есть срока беременности,

Первые две недели беременности являются критическим периодом для внутриутробного развития человека.
Первые две недели беременности являются критическим периодом для внутриутробного развития человека.
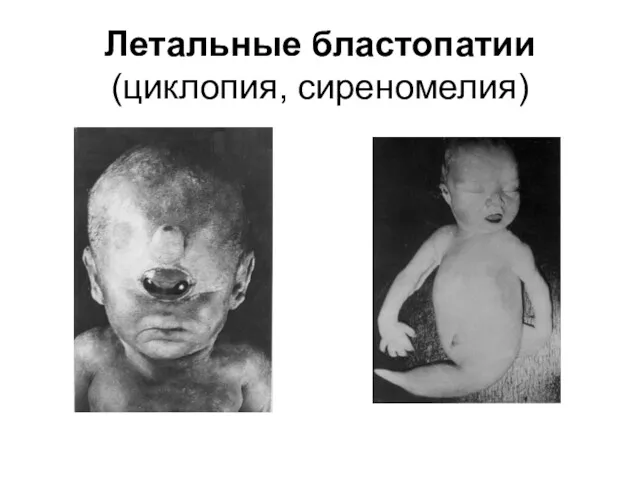
Летальные бластопатии
(циклопия, сиреномелия)
Летальные бластопатии
(циклопия, сиреномелия)

Следующий период активного органогенеза до 12 недели беременности является особенно чувствительным
Следующий период активного органогенеза до 12 недели беременности является особенно чувствительным

К эмбриопатиям относятся такие тяжелые ВПР как дефекты заращения нервной трубки
К эмбриопатиям относятся такие тяжелые ВПР как дефекты заращения нервной трубки
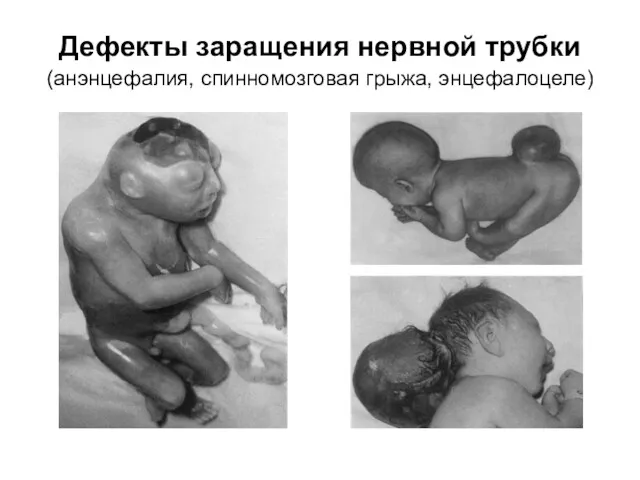
Дефекты заращения нервной трубки (анэнцефалия, спинномозговая грыжа, энцефалоцеле)
Дефекты заращения нервной трубки (анэнцефалия, спинномозговая грыжа, энцефалоцеле)
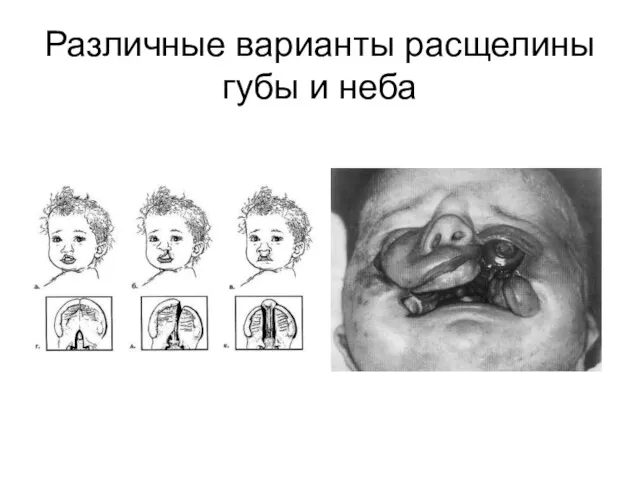
Различные варианты расщелины губы и неба
Различные варианты расщелины губы и неба

Опасность возникновения ВПР под действием тератогенов сохраняется и на следующих стадиях
Опасность возникновения ВПР под действием тератогенов сохраняется и на следующих стадиях
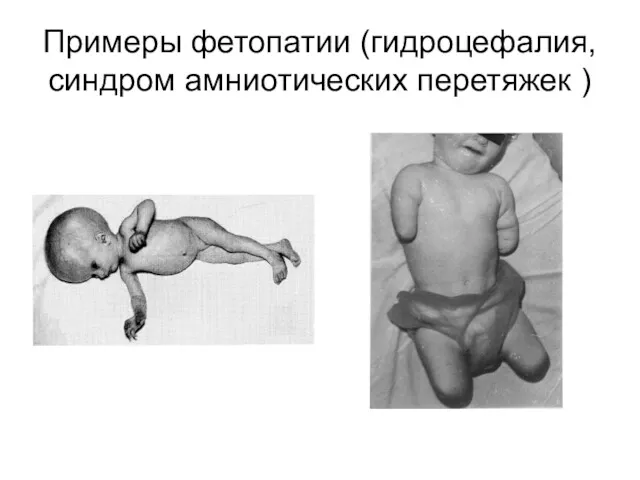
Примеры фетопатии (гидроцефалия, синдром амниотических перетяжек )
Примеры фетопатии (гидроцефалия, синдром амниотических перетяжек )

Причиной развития наследственных болезней является присутствие в половых клетках родителей патологических
Причиной развития наследственных болезней является присутствие в половых клетках родителей патологических

Мутации могут быть геномными, хромосомными и генными. Числовые хромосомные мутации затрагивают
Мутации могут быть геномными, хромосомными и генными. Числовые хромосомные мутации затрагивают

Основная масса зародышей (до 60%) с дисбалансом хромосом погибает в ранний
Основная масса зародышей (до 60%) с дисбалансом хромосом погибает в ранний

У 5% детей, погибших в перинатальном периоде также обнаруживаются хромосомные аномалии.
У 5% детей, погибших в перинатальном периоде также обнаруживаются хромосомные аномалии.

В 34% случаев у детей с хромосомными нарушениями обнаруживаются анеуплоидии по
В 34% случаев у детей с хромосомными нарушениями обнаруживаются анеуплоидии по

Трисомии среди живорожденных описаны лишь для шести хромосом, по остальным хромосомам
Трисомии среди живорожденных описаны лишь для шести хромосом, по остальным хромосомам

Лицевые аномалии при синдроме Дауна
Лицевые аномалии при синдроме Дауна

Больные синдром Патау
Больные синдром Патау

Часто числовые аномалии затрагивают половые хромосомы. Присутствие дополнительной Х-хромосомы у мужчин
Часто числовые аномалии затрагивают половые хромосомы. Присутствие дополнительной Х-хромосомы у мужчин
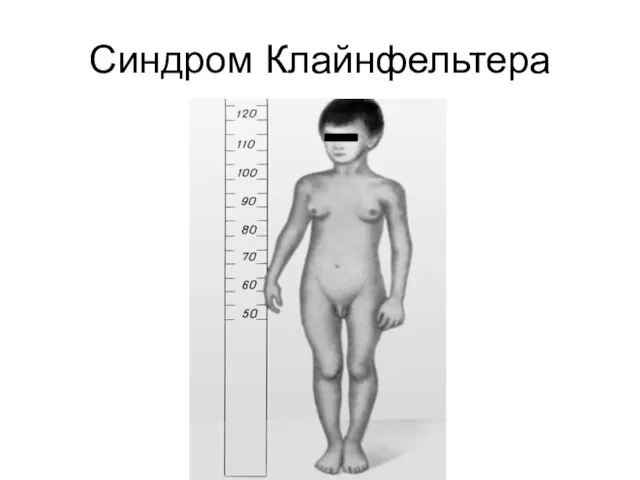
Синдром Клайнфельтера
Синдром Клайнфельтера
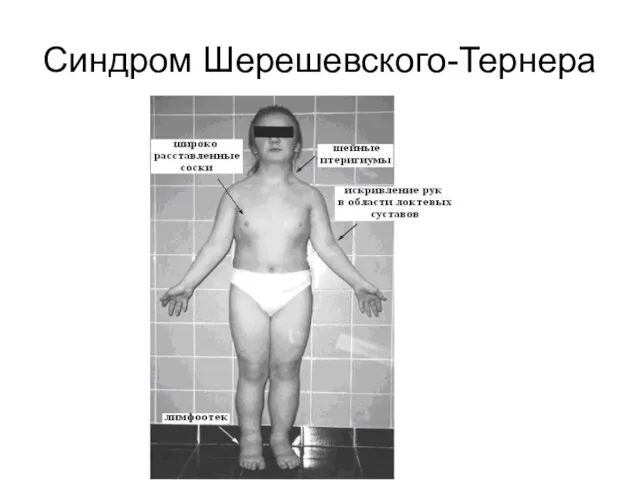
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера

Моногенные болезни обусловлены присутствием мутаций в одном гене.
Следствием мутаций может
Моногенные болезни обусловлены присутствием мутаций в одном гене. Следствием мутаций может

Число моногенных заболеваний достигает 5000. Наиболее частыми из них (1:2-3 до
Число моногенных заболеваний достигает 5000. Наиболее частыми из них (1:2-3 до

Среди моногенных болезней значительный процент составляют ферментопатии, различные формы умственной отсталости,
Среди моногенных болезней значительный процент составляют ферментопатии, различные формы умственной отсталости,

Моногенные заболевания в редких случаях встречаются среди таких нозологических форм, которые
Моногенные заболевания в редких случаях встречаются среди таких нозологических форм, которые

Моногенные варианты заболевания, как правило, отличаются от спорадических форм более тяжелым
Моногенные варианты заболевания, как правило, отличаются от спорадических форм более тяжелым

Несмотря на клиническое многообразие моногенных болезней, можно выделить некоторые общие черты,
Несмотря на клиническое многообразие моногенных болезней, можно выделить некоторые общие черты,

Большинство моногенных болезней распознаются в перинатальном или раннем детском возрасте. Около
Большинство моногенных болезней распознаются в перинатальном или раннем детском возрасте. Около

Некоторые моногенные болезни, такие как спиноцеребеллярные атаксии, миодистрофия Ландузи-Дежерина, хорея Гентингтона,
Некоторые моногенные болезни, такие как спиноцеребеллярные атаксии, миодистрофия Ландузи-Дежерина, хорея Гентингтона,

Типичными чертами многих наследственных заболеваний являются хронический характер и прогредиентность течения
Типичными чертами многих наследственных заболеваний являются хронический характер и прогредиентность течения

При некоторых моногенных заболеваниях выявляются редкие специфические симптомы, проявления которых не
При некоторых моногенных заболеваниях выявляются редкие специфические симптомы, проявления которых не

Внешний вид больных часто столь специфичен, что делает их более похожими
Внешний вид больных часто столь специфичен, что делает их более похожими

Мукополисахаридоз I типа
Мукополисахаридоз I типа

При синдроме Вильямса необычное лицо «эльфа» создается коротким носом, эпикантом, длинным
При синдроме Вильямса необычное лицо «эльфа» создается коротким носом, эпикантом, длинным

Синдром Вильямса
Синдром Вильямса

Черепно-лицевые особенности при синдроме Рассела-Сильвера
Черепно-лицевые особенности при синдроме Рассела-Сильвера

Моногенные заболевания классифицируют по типам наследования, которые в большинстве случаев соответствуют
Моногенные заболевания классифицируют по типам наследования, которые в большинстве случаев соответствуют

Наследование моногенных заболеваний зависит от характера доминирования и нахождения гена в
Наследование моногенных заболеваний зависит от характера доминирования и нахождения гена в
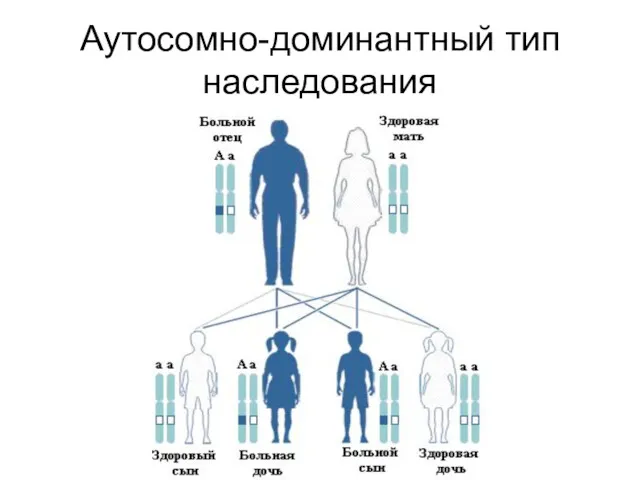
Аутосомно-доминантный тип наследования
Аутосомно-доминантный тип наследования

Особенности
аутосомно-доминантного наследования
Болеют в равной степени мужчины и женщины
Как правило, больные являются
Особенности
аутосомно-доминантного наследования
Болеют в равной степени мужчины и женщины
Как правило, больные являются

Около 50% врожденных пороков развития относятся к аутосомно-доминантным заболеваниям. По аутосомно-доминантному
Около 50% врожденных пороков развития относятся к аутосомно-доминантным заболеваниям. По аутосомно-доминантному

Однако самой многочисленной группой аутосомно-доминантных заболеваний являются наследственные опухолевые синдромы. Их
Однако самой многочисленной группой аутосомно-доминантных заболеваний являются наследственные опухолевые синдромы. Их

Единственным клиническим проявлением наследственных опухолевых синдромов является повышенная вероятность возникновения онкологических
Единственным клиническим проявлением наследственных опухолевых синдромов является повышенная вероятность возникновения онкологических

Наиболее известным аутосомно-доминантным заболеванием является синдром Марфана, при котором у больных
Наиболее известным аутосомно-доминантным заболеванием является синдром Марфана, при котором у больных

Характерными клиническими проявлениями синдрома Марфана являются высокий рост, в сочетании с
Характерными клиническими проявлениями синдрома Марфана являются высокий рост, в сочетании с
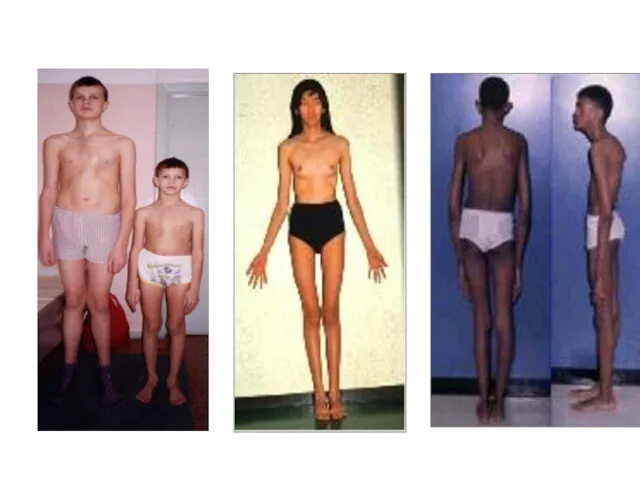

Предполагали, что заболевание обусловлено мутациями в одном из коллагеновых генов. Однако
Предполагали, что заболевание обусловлено мутациями в одном из коллагеновых генов. Однако

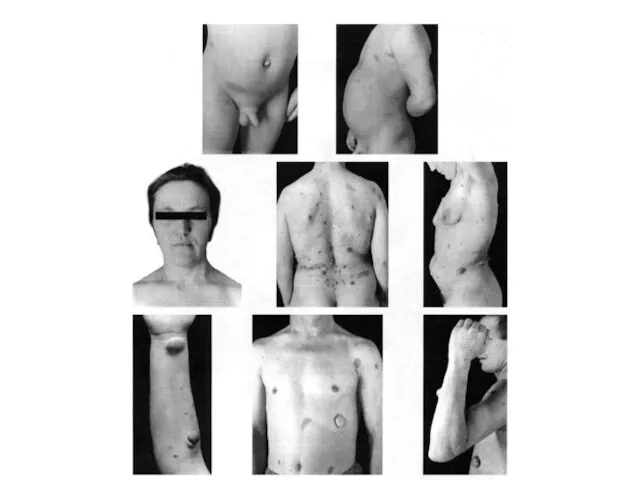
Факоматозы характеризуются сочетанным поражением нервной системы, кожных покровов и внутренних органов.
Факоматозы характеризуются сочетанным поражением нервной системы, кожных покровов и внутренних органов.

Характерными клиническими проявлениями нейрофиброматоза I являются доброкачественные опухоли кожи и подкожной
Характерными клиническими проявлениями нейрофиброматоза I являются доброкачественные опухоли кожи и подкожной

В группу наследственных коллагенопатий, обусловленных мутациями в генах коллагенов и ферментов
В группу наследственных коллагенопатий, обусловленных мутациями в генах коллагенов и ферментов

Коллагены составляют более 30% общей массы белков тела млекопитающих. Разнообразие коллагеновых
Коллагены составляют более 30% общей массы белков тела млекопитающих. Разнообразие коллагеновых

Все коллагеновые альфа-цепи имеют коллагеновый домен, на протяжении которого каждая третья
Все коллагеновые альфа-цепи имеют коллагеновый домен, на протяжении которого каждая третья
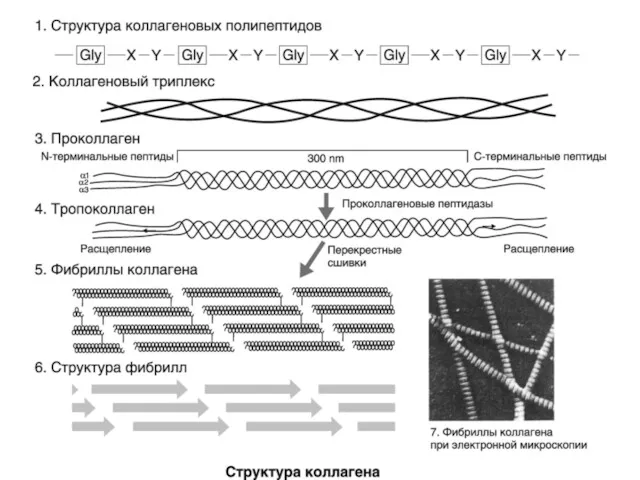

Более 90% коллагеновых волокон образованы мажорными фибриллярными коллагенами I, II и
Более 90% коллагеновых волокон образованы мажорными фибриллярными коллагенами I, II и

Коллаген I типа экспрессируется повсеместно, но особенно обильно представлен в костной
Коллаген I типа экспрессируется повсеместно, но особенно обильно представлен в костной

Клиническая картина несовершенного остеогенеза характеризуется повышенной ломкостью костей и патологическими изменениями
Клиническая картина несовершенного остеогенеза характеризуется повышенной ломкостью костей и патологическими изменениями

Наблюдается высокий клинический полиморфизм заболевания от летальных неонатальных до взрослых форм,
Наблюдается высокий клинический полиморфизм заболевания от летальных неонатальных до взрослых форм,

Неонатальная форма несовершенного остеогенеза
Неонатальная форма несовершенного остеогенеза
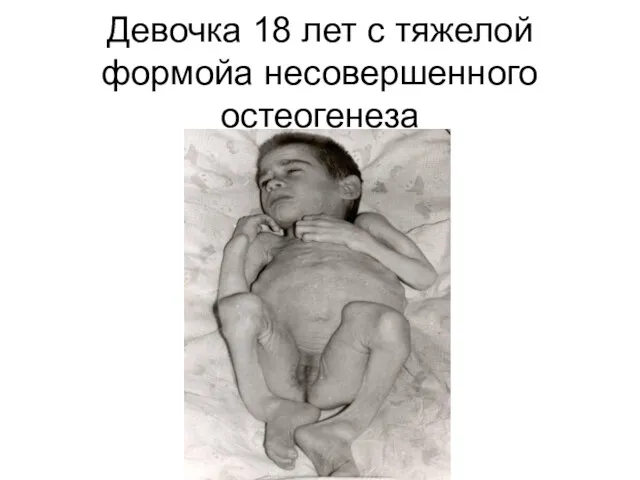
Девочка 18 лет с тяжелой формойа несовершенного остеогенеза
Девочка 18 лет с тяжелой формойа несовершенного остеогенеза

Оказалось, что при тяжелых формах заболевания часто обнаруживаются миссенс-мутации, изменяющие положения
Оказалось, что при тяжелых формах заболевания часто обнаруживаются миссенс-мутации, изменяющие положения

При легких формах частыми являются нонсенс-мутации.
При этом мутантная альфа-цепь деградирует
При легких формах частыми являются нонсенс-мутации. При этом мутантная альфа-цепь деградирует

Коллаген II типа является мажорным хрящевым коллагеном и составляет основу стекловидного
Коллаген II типа является мажорным хрящевым коллагеном и составляет основу стекловидного

Спондилоепиметафизарная дисплазия
Спондилоепиметафизарная дисплазия
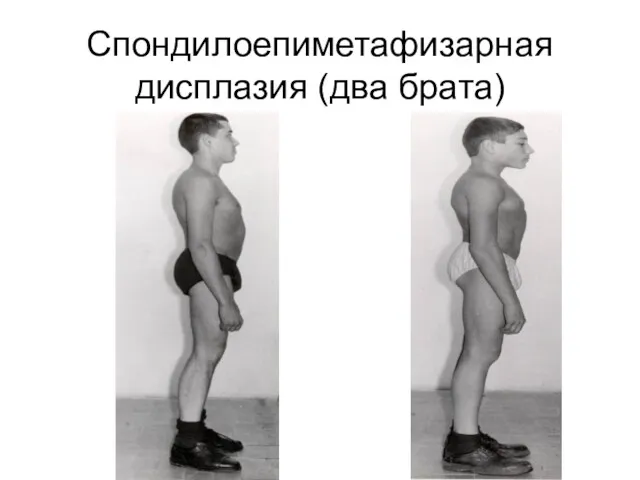
Спондилоепиметафизарная дисплазия (два брата)
Спондилоепиметафизарная дисплазия (два брата)

Синдром Элерса-Данло характеризуется гиперрастяжимостью и истончением кожи, гипермобильностью суставов, скелетными аномалиями,
Синдром Элерса-Данло характеризуется гиперрастяжимостью и истончением кожи, гипермобильностью суставов, скелетными аномалиями,

Клинические проявления синдрома Элерса-Данло
Клинические проявления синдрома Элерса-Данло

Скелетные аномалии при синдроме Элерса-Данло
Скелетные аномалии при синдроме Элерса-Данло

Семейный случай синдрома Элерса-Данло
Семейный случай синдрома Элерса-Данло

Описано около 10 наследственных вариантов синдрома Элерса-Данло. Классические формы заболевания обусловлены
Описано около 10 наследственных вариантов синдрома Элерса-Данло. Классические формы заболевания обусловлены

Самая тяжелая «артериальная» форма синдрома Элерса-Данло, которая может сопровождаться разрывами артерий
Самая тяжелая «артериальная» форма синдрома Элерса-Данло, которая может сопровождаться разрывами артерий
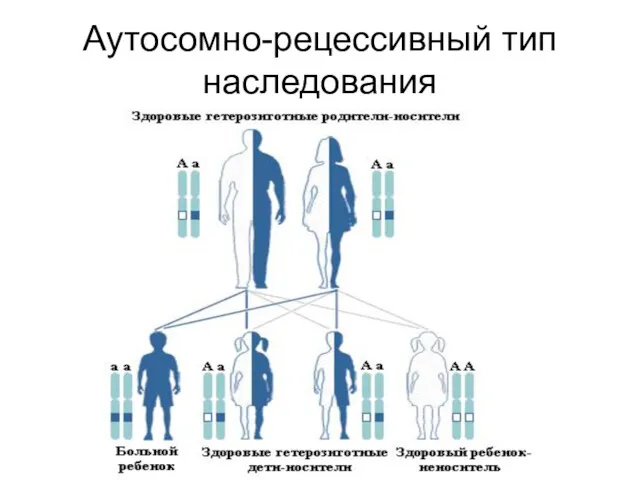
Аутосомно-рецессивный тип наследования
Аутосомно-рецессивный тип наследования

Особенности
аутосомно-рецессивного наследования
Больные дети являются гомозиготными носителями мутаций
Они рождаются с вероятностью 25%
Особенности
аутосомно-рецессивного наследования
Больные дети являются гомозиготными носителями мутаций
Они рождаются с вероятностью 25%

Частоты гетерозигот по рецессивным заболеваниям с распространенностью:
1 на 2-20 000 –1:20-70,
Частоты гетерозигот по рецессивным заболеваниям с распространенностью: 1 на 2-20 000 –1:20-70,

Поэтому выдвигавшиеся в начале XX века евгенические предложения по стерилизации больных
Поэтому выдвигавшиеся в начале XX века евгенические предложения по стерилизации больных

По аутосомно-рецессивному типу наследуются болезни обмена (НБО) – одна из наиболее
По аутосомно-рецессивному типу наследуются болезни обмена (НБО) – одна из наиболее

НБО обусловлены нарушением каталитической функции различных ферментов, участвующих в обмене аминокислот,
НБО обусловлены нарушением каталитической функции различных ферментов, участвующих в обмене аминокислот,

Подобные нарушения часто сопровождаются накоплением веществ, предшествующих ферментативному блоку, и дефицитом
Подобные нарушения часто сопровождаются накоплением веществ, предшествующих ферментативному блоку, и дефицитом

Общими нарушениями при наследственных дефектах обмена аминокислот являются аминоацидурия (выделение аминокислот
Общими нарушениями при наследственных дефектах обмена аминокислот являются аминоацидурия (выделение аминокислот
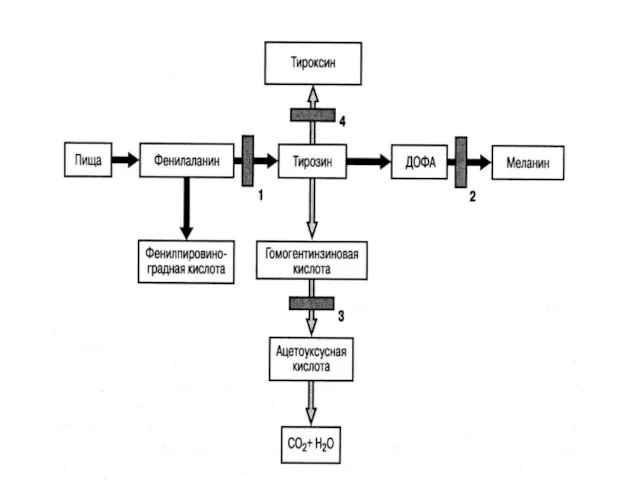

Гиперфенилаланинемии (ГФА) – это группа генетически гетерогенных аутосомно-рецессивных заболеваний, обусловленных нарушением
Гиперфенилаланинемии (ГФА) – это группа генетически гетерогенных аутосомно-рецессивных заболеваний, обусловленных нарушением

В основе патогенеза ГФА лежит накопление в крови фенилаланина (незаменимой аминокислоты,
В основе патогенеза ГФА лежит накопление в крови фенилаланина (незаменимой аминокислоты,

Фенилкетонурия (ФКУ), наиболее частая и злокачественная форма ГФА. Заболевание обусловлено наследственной
Фенилкетонурия (ФКУ), наиболее частая и злокачественная форма ГФА. Заболевание обусловлено наследственной

Ведущим симптомом болезни является отставание умственного развития – олигофрения. Уже с
Ведущим симптомом болезни является отставание умственного развития – олигофрения. Уже с

Лечение больных заключается в исключении из питания фенилаланина путем применения специфической
Лечение больных заключается в исключении из питания фенилаланина путем применения специфической

Успех лечения зависит от того, насколько рано поставлен диагноз.
Поэтому всем
Успех лечения зависит от того, насколько рано поставлен диагноз. Поэтому всем

Больные фенилкетонурией, выявленные по неонатальному скринигу
Больные фенилкетонурией, выявленные по неонатальному скринигу

В России имеется мажорная миссенс-мутация R408W в гене фенилаланингидроксилазы (PAH), частота
В России имеется мажорная миссенс-мутация R408W в гене фенилаланингидроксилазы (PAH), частота
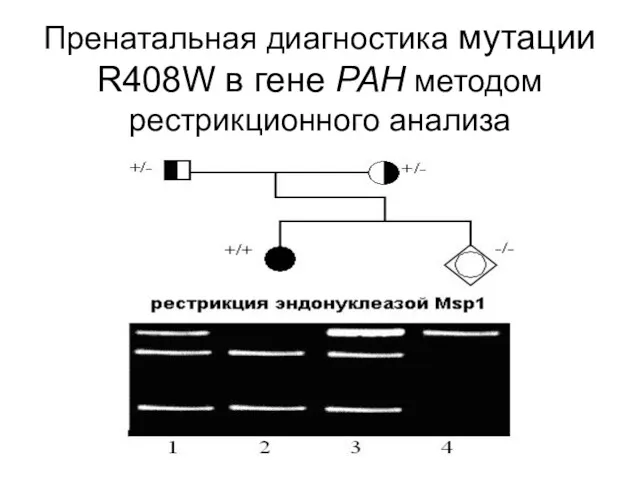
Пренатальная диагностика мутации R408W в гене PAH методом рестрикционного анализа
Пренатальная диагностика мутации R408W в гене PAH методом рестрикционного анализа

Наиболее распространенным аутосомно-рецессивным заболеванием среди европейцев является муковисцидоз. Его частота в
Наиболее распространенным аутосомно-рецессивным заболеванием среди европейцев является муковисцидоз. Его частота в

Молекулярной основой патогенеза муковисцидоза является нарушение работы хлорного канала, локализованного на
Молекулярной основой патогенеза муковисцидоза является нарушение работы хлорного канала, локализованного на

Самой распространенной мутаций в гене муковисцидоза (CFTR) является delF508 – делеция
Самой распространенной мутаций в гене муковисцидоза (CFTR) является delF508 – делеция

Присутствие мутации delF508 у больных и гетерозиготных носителей легко определить методом
Присутствие мутации delF508 у больных и гетерозиготных носителей легко определить методом
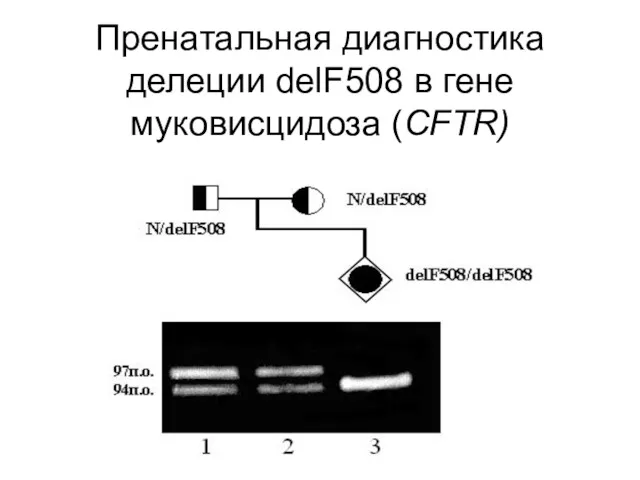
Пренатальная диагностика делеции delF508 в гене муковисцидоза (CFTR)
Пренатальная диагностика делеции delF508 в гене муковисцидоза (CFTR)

Спинальная мышечная атрофия I
Спинальная мышечная атрофия I

Динамика почерка больной ГЛД на фоне лечения в течение 36 лет
Динамика почерка больной ГЛД на фоне лечения в течение 36 лет
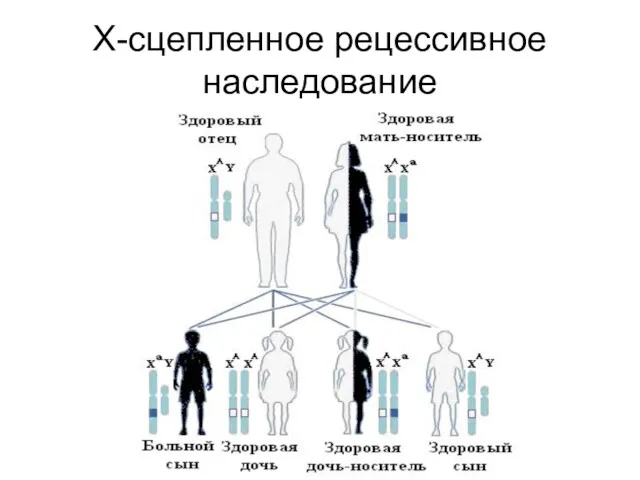
Х-сцепленное рецессивное наследование
Х-сцепленное рецессивное наследование
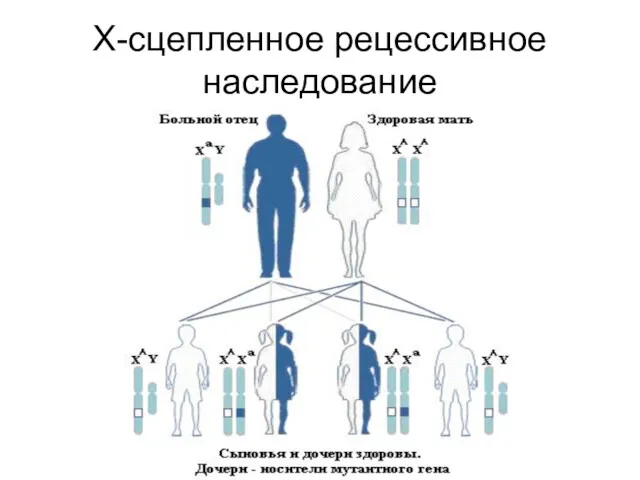
Х-сцепленное рецессивное наследование
Х-сцепленное рецессивное наследование

Особенности Х-сцепленного рецессивного наследования
Болеют только мальчики
Оба родителя здоровы, но мать несет
Особенности Х-сцепленного рецессивного наследования
Болеют только мальчики
Оба родителя здоровы, но мать несет

Наиболее известными Х-сцепленными рецессивными заболеваниями являются гемофилия А и В, миодистрофия
Наиболее известными Х-сцепленными рецессивными заболеваниями являются гемофилия А и В, миодистрофия

Синдром Мартина Белл
Синдром Мартина Белл
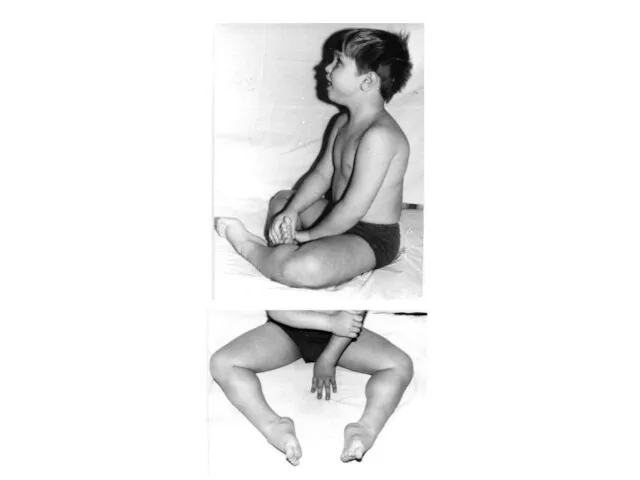

Основным продуктом гена миодистрофии Дюшенна (DMD) является стержневидный белок дистрофин, располагающейся
Основным продуктом гена миодистрофии Дюшенна (DMD) является стержневидный белок дистрофин, располагающейся

Дистрофин – это полифункциональный белок, обеспечивающий поддержание целостности мембраны мышечного волокна
Дистрофин – это полифункциональный белок, обеспечивающий поддержание целостности мембраны мышечного волокна
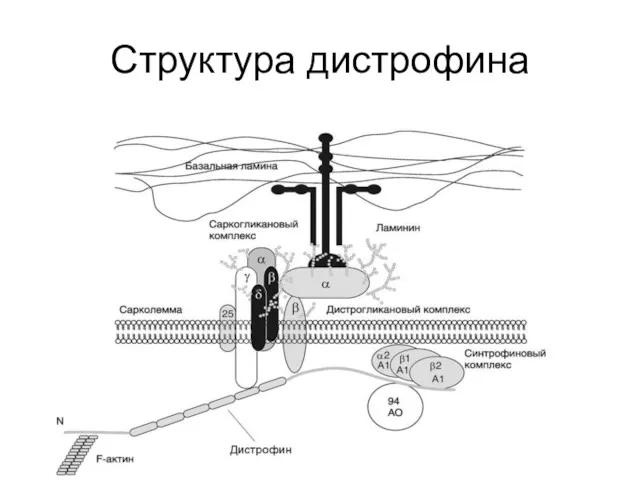
Структура дистрофина
Структура дистрофина

В большинстве случаев у больных диагностируются протяженные внутригенные делеции в гене
В большинстве случаев у больных диагностируются протяженные внутригенные делеции в гене

Диагностика делеций в гене DMD осуществляется методом мультиплексной ПЦР, при которой
Диагностика делеций в гене DMD осуществляется методом мультиплексной ПЦР, при которой
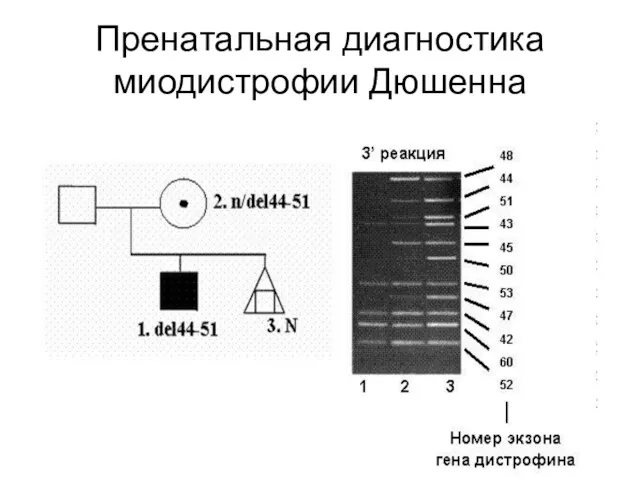
Пренатальная диагностика миодистрофии Дюшенна
Пренатальная диагностика миодистрофии Дюшенна

Некоторые моногенные заболевания не подчиняется законам Менделя. Это митохондриальные заболевания, болезни
Некоторые моногенные заболевания не подчиняется законам Менделя. Это митохондриальные заболевания, болезни
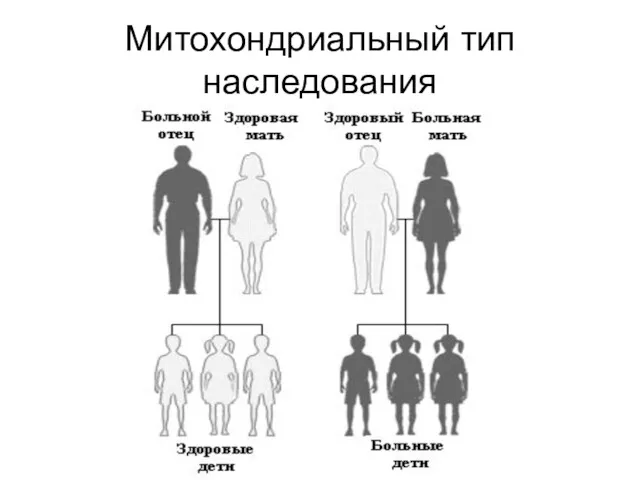
Митохондриальный тип наследования
Митохондриальный тип наследования

К Мт-болезням относятся синдром Лебера (атрофия зрительного нерва), MELAS-синдром (лактоацидоз с
К Мт-болезням относятся синдром Лебера (атрофия зрительного нерва), MELAS-синдром (лактоацидоз с

Многофакторные или комплексные заболевания обусловлены комбинированным действием неблагоприятных средовых и генетических
Многофакторные или комплексные заболевания обусловлены комбинированным действием неблагоприятных средовых и генетических

К ним относятся социально-значимыме хронические болезни, такие как сердечно-сосудистая патология, диабет,
К ним относятся социально-значимыме хронические болезни, такие как сердечно-сосудистая патология, диабет,

В качестве генетических факторов риска многофакторной патологии рассматривают широко распространенные полиморфные
В качестве генетических факторов риска многофакторной патологии рассматривают широко распространенные полиморфные

Поиск генов-кандидатов, формирующих «генную сеть» многофакторного заболевания, осуществляют, исходя из знаний
Поиск генов-кандидатов, формирующих «генную сеть» многофакторного заболевания, осуществляют, исходя из знаний

Что мы знаем о заболевании? Какие метаболические циклы дефектны при тех
Что мы знаем о заболевании? Какие метаболические циклы дефектны при тех

Есть ли там широко распространенные среди населения (полиморфные) аллели, влияющие на
Есть ли там широко распространенные среди населения (полиморфные) аллели, влияющие на

Для того чтобы определить, является ли полиморфный аллель генетическим фактором риска,
Для того чтобы определить, является ли полиморфный аллель генетическим фактором риска,

Только в тех случаях, когда уровни полиморфизма среди больных оказываются достоверно
Только в тех случаях, когда уровни полиморфизма среди больных оказываются достоверно

Идентификация генетических факторов риска и разработка индивидуальных профилактических мероприятий составляют стратегическую
Идентификация генетических факторов риска и разработка индивидуальных профилактических мероприятий составляют стратегическую

Фармакогенетика – это раздел медицинской генетики, изучающий влияние наследственной конституции на
Фармакогенетика – это раздел медицинской генетики, изучающий влияние наследственной конституции на

По разным оценкам вклад генетических составляющих в вариабельность реакции на лекарственные
По разным оценкам вклад генетических составляющих в вариабельность реакции на лекарственные

В настоящее время в рамках некоторых международных проектов (PharmacoGenetics for Every
В настоящее время в рамках некоторых международных проектов (PharmacoGenetics for Every

Описан ряд наследственных болезней обмена, ведущих к медикаментозным идиосинкразиям
Описан ряд наследственных болезней обмена, ведущих к медикаментозным идиосинкразиям

Мутации в гене псевдохолинэстеразы препятствуют гидролизу используемого в анестезиологии препарата суксаметония.
Мутации в гене псевдохолинэстеразы препятствуют гидролизу используемого в анестезиологии препарата суксаметония.

В гене Г-6-ФДГ идентифицированы мутации, ассоциированные с различными формами гемолитической анемии.
В гене Г-6-ФДГ идентифицированы мутации, ассоциированные с различными формами гемолитической анемии.

Прием барбитуратов, сульфаниламидов, некоторых противосудорожных препаратов и антибиотиков может привести к
Прием барбитуратов, сульфаниламидов, некоторых противосудорожных препаратов и антибиотиков может привести к

Ее клиническими проявлениями являются острые боли в животе, красный цвет мочи,
Ее клиническими проявлениями являются острые боли в животе, красный цвет мочи,

Наследуется заболевание по аутосомно-доминантному типу с неполной пенетрантностью, его частота в
Наследуется заболевание по аутосомно-доминантному типу с неполной пенетрантностью, его частота в

При некоторых наследственных заболеваниях может наблюдаться необычная реакция на определенные лекарственные
При некоторых наследственных заболеваниях может наблюдаться необычная реакция на определенные лекарственные

Заболевание обусловлено ускоренным синтезом мочевой кислоты с одновременным снижением ее выведения
Заболевание обусловлено ускоренным синтезом мочевой кислоты с одновременным снижением ее выведения

Некоторые диуретики (хлортиазид, фуросемид) могут ускорить и резко усилить клинические проявления
Некоторые диуретики (хлортиазид, фуросемид) могут ускорить и резко усилить клинические проявления

Все рассмотренные выше примеры касались достаточно редких моногенных болезней. Однако различная
Все рассмотренные выше примеры касались достаточно редких моногенных болезней. Однако различная

В большинстве случаев она связана не с наследственными заболеваниями, а с
В большинстве случаев она связана не с наследственными заболеваниями, а с

Одним из наиболее ярких примеров подобного рода является влияние скорости ацетилирования,
Одним из наиболее ярких примеров подобного рода является влияние скорости ацетилирования,

В гене NAT2 имеются полиморфные аллели, влияющие на активность соответствующего фермента.
В гене NAT2 имеются полиморфные аллели, влияющие на активность соответствующего фермента.

В европейских популяциях соотношение между этими двумя группами населения примерно одинаково,
В европейских популяциях соотношение между этими двумя группами населения примерно одинаково,

«Медленные» ацетиляторы в большей степени склонны к интоксикации. В частности, при
«Медленные» ацетиляторы в большей степени склонны к интоксикации. В частности, при

Его накопление сопровождается дефицитом нейротропных витаминов группы В. Поэтому при терапии
Его накопление сопровождается дефицитом нейротропных витаминов группы В. Поэтому при терапии

«Медленные» ацетиляторы в большей степени склонны к развитию гемолитической анемии при
«Медленные» ацетиляторы в большей степени склонны к развитию гемолитической анемии при

Дозы изониазида могут быть снижены, если больные относятся к группе «медленных»
Дозы изониазида могут быть снижены, если больные относятся к группе «медленных»

Ключевая роль в метаболизме многих лекарственных препаратов принадлежит цитохромам P450. Их
Ключевая роль в метаболизме многих лекарственных препаратов принадлежит цитохромам P450. Их

Разберем более подробно связь цитохромов P450 с лекарственным метаболизмом на примере
Разберем более подробно связь цитохромов P450 с лекарственным метаболизмом на примере

Этот цитохром непосредственно взаимодействует с S-варфарином, который широко используется при лечении
Этот цитохром непосредственно взаимодействует с S-варфарином, который широко используется при лечении

Успех лечения зависит от выбора дозы препарата и длительности курса. С
Успех лечения зависит от выбора дозы препарата и длительности курса. С

В гене CYP2C9 идентифицированы два функциональных полиморфизма, влияющие на метаболическую активность
В гене CYP2C9 идентифицированы два функциональных полиморфизма, влияющие на метаболическую активность

В отечественных популяциях суммарная частота носителей минорных аллелей гена CYP2C9 достигает
В отечественных популяциях суммарная частота носителей минорных аллелей гена CYP2C9 достигает

Больные с низкой активностью метаболизатора имеют большую вероятность геморрагических осложнений при
Больные с низкой активностью метаболизатора имеют большую вероятность геморрагических осложнений при

Значимыми с фармакологической точки зрения являются некоторые полиморфные аллели в других
Значимыми с фармакологической точки зрения являются некоторые полиморфные аллели в других

Кроме того, в биотрансформации лекарственных препаратов принимают участие многие другие ферменты
Кроме того, в биотрансформации лекарственных препаратов принимают участие многие другие ферменты

Одной из ведущих проблем современной фармакогенетики является разработка схем терапии различных
Одной из ведущих проблем современной фармакогенетики является разработка схем терапии различных

Описание различных этнических групп по специфическим профилям полиморфизмов генов лекарственного метаболизма
Описание различных этнических групп по специфическим профилям полиморфизмов генов лекарственного метаболизма

Лечение больных с врожденной и наследственной патологией в основном носит симптоматический
Лечение больных с врожденной и наследственной патологией в основном носит симптоматический

При этом могут быть использованы самые разнообразные методы терапии – медикаментозные,
При этом могут быть использованы самые разнообразные методы терапии – медикаментозные,

Результаты симптоматического лечения, обычно, не очень продолжительны, требуется многократное повторение лечебных
Результаты симптоматического лечения, обычно, не очень продолжительны, требуется многократное повторение лечебных

Необходимыми элементами воспитания больных синдромом Дауна являются длительные упражнения по развитию
Необходимыми элементами воспитания больных синдромом Дауна являются длительные упражнения по развитию

Наряду с этим, желательно применение препаратов нооторопного действия, фолатов, витаминотераприя и
Наряду с этим, желательно применение препаратов нооторопного действия, фолатов, витаминотераприя и

При синдроме Шерешевского-Тернера применяются гормональная терапия для стимуляции роста и препараты
При синдроме Шерешевского-Тернера применяются гормональная терапия для стимуляции роста и препараты

При наличии гинекомастии у мужчин с синдромом Клайнфельтера производят удаление молочных
При наличии гинекомастии у мужчин с синдромом Клайнфельтера производят удаление молочных

Патогенетическое лечение наследственных болезней направлено на коррекцию биохимических и физиологических процессов,
Патогенетическое лечение наследственных болезней направлено на коррекцию биохимических и физиологических процессов,

Самым простым методом патогенетического лечения является диетотерапия. Но она эффективна лишь
Самым простым методом патогенетического лечения является диетотерапия. Но она эффективна лишь

Наиболее эффективным способом терапевтической коррекции наследственных энзимопатий является введение недостающего фермента
Наиболее эффективным способом терапевтической коррекции наследственных энзимопатий является введение недостающего фермента

Основными проблемами при этом являются: (1) получение в достаточном количестве чистого
Основными проблемами при этом являются: (1) получение в достаточном количестве чистого

Большого успеха удалось достичь при лечении с помощью заместительной ферментотерапии болезни
Большого успеха удалось достичь при лечении с помощью заместительной ферментотерапии болезни

Хорошие результаты получены при заместительной ферментотерапии мукополисахаридозов, особенно в тех случаях,
Хорошие результаты получены при заместительной ферментотерапии мукополисахаридозов, особенно в тех случаях,

При заболеваниях, обусловленных дефицитом конечного продукта патологической метаболической цепи, лечение направлено
При заболеваниях, обусловленных дефицитом конечного продукта патологической метаболической цепи, лечение направлено

Примером является лечение врожденного гипотиреоза тироксином, своевременное назначение которого полностью предотвращает
Примером является лечение врожденного гипотиреоза тироксином, своевременное назначение которого полностью предотвращает

Наиболее перспективным способом этиологического лечения наследственных заболеваний является исправление генетических дефектов,
Наиболее перспективным способом этиологического лечения наследственных заболеваний является исправление генетических дефектов,

Количество генотерапевтических проектов, находящихся на стадии клинических испытаний, в США уже
Количество генотерапевтических проектов, находящихся на стадии клинических испытаний, в США уже
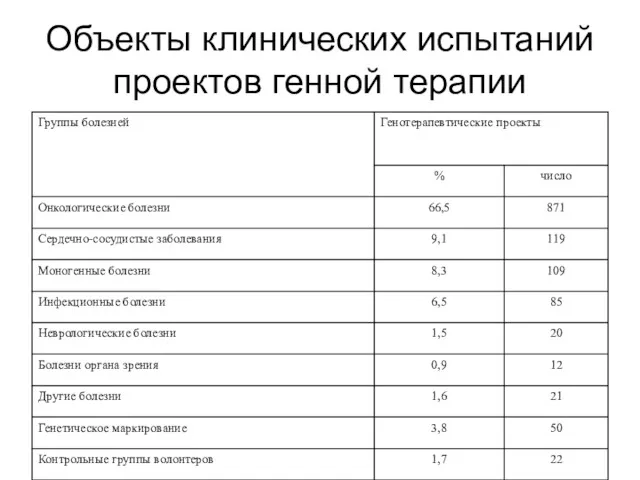
Объекты клинических испытаний проектов генной терапии
Объекты клинических испытаний проектов генной терапии

Программы генной терапии оказались наиболее успешны при лечении некоторых наследственных иммунодефицитов
Программы генной терапии оказались наиболее успешны при лечении некоторых наследственных иммунодефицитов

Однако в настоящее время нет ни одного генотерапевтического проекта, который имел
Однако в настоящее время нет ни одного генотерапевтического проекта, который имел

Таким образом, генотерапия остается одним из наиболее привлекательных и перспективных направлений
Таким образом, генотерапия остается одним из наиболее привлекательных и перспективных направлений
 Injury of genitourinary organs
Injury of genitourinary organs Структура детской поликлиники. Участковый принцип обслуживания детей
Структура детской поликлиники. Участковый принцип обслуживания детей Составление схем диспансерного наблюдения у курируемых хронических больных
Составление схем диспансерного наблюдения у курируемых хронических больных Проблема обеспеченности медицинской помощью жителей Бурятии
Проблема обеспеченности медицинской помощью жителей Бурятии О переводе государственных услуг в сфере здравоохранения в электронный вид
О переводе государственных услуг в сфере здравоохранения в электронный вид Паллиативный уход
Паллиативный уход Обучающий семинар Нефро-лиги. Пациент и лекарственное обеспечение
Обучающий семинар Нефро-лиги. Пациент и лекарственное обеспечение Травмы сердца
Травмы сердца Отравляющие и высокотоксичные вещества общеядовитого действия
Отравляющие и высокотоксичные вещества общеядовитого действия Гломерулонефрит и беременность
Гломерулонефрит и беременность Роль и значение анестезиологии, реанимации и интенсивной терапии в современной медицине
Роль и значение анестезиологии, реанимации и интенсивной терапии в современной медицине Перекрестный прикус
Перекрестный прикус Гигиенические требования, предъявляемые к пищевым продуктам
Гигиенические требования, предъявляемые к пищевым продуктам Чрескожные коронарные вмешательства
Чрескожные коронарные вмешательства Простые советы по профилактике онкологических заболеваний
Простые советы по профилактике онкологических заболеваний Дитячі інфекційні хвороби
Дитячі інфекційні хвороби Репродуктивті денсаулық және мінез құлық. Сырқаттылықты жеке және жалпы тіркеу. Медициналық сақтандыру
Репродуктивті денсаулық және мінез құлық. Сырқаттылықты жеке және жалпы тіркеу. Медициналық сақтандыру Акушерство. История развития акушерства
Акушерство. История развития акушерства Антропометриялық көрсеткіштерді анықтау
Антропометриялық көрсеткіштерді анықтау 20231028_nezhelatelnye_fizicheskie_uprazhneniya_na_urokah_fizicheskoy_kultury_0
20231028_nezhelatelnye_fizicheskie_uprazhneniya_na_urokah_fizicheskoy_kultury_0 Осложнения, возникающие во время и после удаления зуба
Осложнения, возникающие во время и после удаления зуба Оценка эффективности первого этапа диспансеризации за 2017 год
Оценка эффективности первого этапа диспансеризации за 2017 год Амбулаторные операции в полости рта
Амбулаторные операции в полости рта Дифференциальная диагностика синдрома крупа у детей
Дифференциальная диагностика синдрома крупа у детей Основи медичних знань. Сучасні уявлення про здоров'я людини
Основи медичних знань. Сучасні уявлення про здоров'я людини Аллергия. Аллергические реакции
Аллергия. Аллергические реакции Бешенство
Бешенство Расстройство кровообращения, лимфообращения и содержания тканевой жидкости у животных
Расстройство кровообращения, лимфообращения и содержания тканевой жидкости у животных