- Митохондриальные болезни или опять во всем виноваты женщины

Содержание
- 2. Содержимое клетки
- 4. Немного истории 1949 год B.Ephrussi с соавторами открывает "цитоплазматическую" наследованную "малую" мутацию у дрожжей (факультативные организмы),
- 5. Немного истории 1962 год - R.Luft c сотрудниками впервые описал первую болезнь человека, причиной которой были
- 6. ЭНДОСИМБИОНТЫ митохондрии и хлоропласты (с ними связан процесс трансдукции энергии внутрь кпетки) являются прямыми потомками свободноживущих
- 7. ЭНДОСИМБИОНТЫ принесли в клетку бактериальный геном, остатки которого продолжают существовать сегодня в виде МИОХОНДРИАЛЬНОЙ ДНК (мхДНК)
- 8. ЭНДОСИМБИОТИЧЕСКАЯ ГИПОТЕЗА была выдвинута 100 лет назад - однако факты, подтверждающие эту теорию были получены лишь
- 9. Происхождение органелл клетки Определение происхождения органелл осуществляют по рРНК Сравнение гомологичных последовательностей рРНК хлоропластов и митохондрий
- 11. Происхождение органелл клетки Определение происхождения органелл осуществляют по рРНК Сравнение гомологичных последовательностей рРНК хлоропластов и митохондрий
- 12. Происхождение органелл клетки Гены рРНК расположены среди нескольких генов, встречающихся как в мхДНК, хпДНК, так и
- 13. Происхождение органелл клетки Хлоропласты и митохондрии пришли из совершенно различных групп эубактерий: класс ХЛОРОПЛАСТОВ из группы
- 14. Строение митохондрий Общий вид органеллы
- 15. Строение митохондрий
- 16. Строение митохондрий
- 17. Строение митохондрий
- 18. Строение митохондрий Электронное фото
- 19. Строение митохондрий Электронная фотография
- 20. Питер МИТЧЕЛЛ Нобелевская премия 1978 года
- 21. Строение митохондрий Устройство крист
- 22. Механизм генерации энергии
- 23. Устройство дыхательной цепи
- 24. Количество мхДНК в клетках одна митохондрия содержит около 10 молекул мтДНК число копий мтДНК в цитоплазме
- 25. В одной клетке от 100 до 1000 митохондрий
- 26. Строение ДНК митохондрий Митохондриальный геном человека был идентифицирован в 1960 году и расшифрован группой Frederick Sanger
- 27. Геном митохондриальной ДНК
- 30. Митохондриальный геном Человека и локализация некоторых точечных мутаций и типичных делеций Гены, кодирующие: ND1-6 субъединицы комплекса
- 31. Геном митохондриальной ДНК 16.569 основных нуклеотидных пар на них закодировано 37 генов 13 гена полипептидов 22
- 32. Отличие генома митохондрий Общий принцип построения геномов митохондрий - максимальная структурная компактность при максимальной информационной нагруженности.
- 33. Отличие генома митохондрий Генетический код митохондрий позвоночных (человека) 22 антикодона тРНК «узнают» все 60 кодонов мРНК.
- 34. Отличие генома митохондрий Экономичность генома достигается благодаря 1 отсутствию интронов в структурных генах, 2 сведению к
- 35. Отличие генома митохондрий Причина отличия митохондриального кода от ядерного Для нейтрализации АФК в клетке работает несколько
- 36. Наследование митохондриальных болезней происходит только по материнской линии Митохондрии материнской яйцеклетки содержат 100 000 копий мхДНК,
- 37. Оплодотворение яйцеклетки
- 38. Опять во всем виновата Ева ?
- 39. Болезнь первая - синдром Luft 1960 год Rolf Luft и Lars Ernster впервые был опубликован клинический
- 40. Частота встречаемости болезней митохондрий в Англии - 1 на 5 000 человек то есть в Томске
- 41. Причина митохондриальных болезней Глубокое вовлечение в процесс дефекта митохондриальной продукции энергии АТФ. Было установлено, что процессы
- 42. Типичные симптомы мх болезней Известно около 50 митохондриальных болезней. В их клинике доминируют поражения центральной нервной
- 43. Клинический диагноз митохондриальной болезни основан 1. Биохимических признаках (выраженный молочнокислый ацидоз и дефицит работы дыхательной цепи)
- 44. Подход к классификации митохондриальных болезней 1. Смысловые замены в структурных генах; 2. Мутации в генах рРНК
- 45. ВИДЫ МУТАЦИЙ мхДНК и вызванные этими нарушениями митохондриальные болезни
- 46. Точковые мутации и их последствия
- 47. Точковые мутации Болезнь Лебера Болезнь Лебера Leber's hereditary optic neuropathy - LHON наследственная нейропатия зрительных нервов
- 48. Точковые мутации Основной исследователь – Douglas Wallace и сотрудники (1988)
- 49. Точковые мутации Болезнь Лебера ПРИЧИНЫ РАЗВиТИЯ БОЛЕЗНИ Данная мутация выбивает один кодон в одном из триплетов
- 50. Точковые мутации Болезнь Лебера Болезнь чаще поражает мужчин (в соотношении 4: 1,3: Впервые причинно-следственная связь между
- 51. Точковые мутации Болезнь Лебера Клинические признаки при данных мутациях различной локализации существенно не различаются – поэтому
- 52. Точковые мутации Болезнь Лебера Со времени открытия гиганских делеций и точечных мутаций в мхДНК были описаны
- 53. Точковые мутации Пигментный ретинит Пигментный ретинит NARP ("neuropathy, ataxia, retinitis, pigmentosa" (нейропатия, атаксия и пигментный ретинит)
- 54. Точковые мутации Пигментный ретинит ПРИЧИНА Наличие точечной мутации гена 6-ой субъединицы Н+АТФазы, что приводит к замене
- 55. ДЕЛЕЦИИ мхДНК (ВЫПАДЕНИЕ ЕНЕТИЧЕСКОЙ ИНФОРМАЦИИ) и их последствия
- 56. Делеции мхДНК 1 РЕО - "progressive external ophthalmoplegia" (прогрессирующей экстраофтальмоплегией) 2 Синдром KSS Kearns-Sayre Syndrome (мультисистемные
- 57. Делеции мхДНК ОСНОВНОЙ ИССЛЕДОВАТЕЛЬ Ian Holt и его коллеги идентифицировали пациентов с клиническими биохимическими и морфологическими
- 58. Делеции мхДНК ПРИЧИНЫ РАЗВИТИЯ БОЛЕЗНИ Крупные делеции мхДНК обычно БЛОКИРУЮТ ТРАНСКРИПЦИЮ всех митохондриальных генов дыхательной цепи
- 59. Делеции мх ДНК ОБЩИЕ ПРИЗНАКИ одиночные проявления или являющихся частью мультисистемных нарушений
- 60. Делеции ДНК PEO (прогрессирующая экстраофтальмоплегия) Клинически проявляется параличом экстраокулярных мускул, включая птоз Синдром Pearson фатальные гематологические
- 61. Делеции мхДНК Синдром Kearns-Sayre ("Kearns-Sayre syndrome" - KSS) Клинически проявляется в развитии: - хронической наружной офтальмоплегией
- 62. Делеции мх ДНК ОБЩИЕ ПРИЗНАКИ эти мутации потомством не наследуются, так как Ближайшие родственники ни матери,
- 63. Делеции мх ДНК ОБЩИЕ ПРИЗНАКИ при незначительном содержании делетированной мхДНК в тканях организма клинические проявления болезни
- 64. Делеции мх ДНК фенотипические проявления одного и того же дефекта делеции мхДНК в ЗАВИСИМОСТИ ОТ РАСПРЕДЕЛЕНИЯ
- 65. ДУПЛИКАЦИЯ мхДНК и их последствия
- 66. Дупликация мх ДНК ПРИЧИНЫ РАЗВИТИЯ БОЛЕЗНИ две формы мхДНК – одна полноразмерная молекула ДНК и одна
- 67. Дупликация мх ДНК КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ - митохондриальные миопатии - множественные поражения других органов и систем (повреждение
- 68. ОСОБЕННОСТИ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
- 69. 1 НИЗКИЕ ПОРОГИ ДИСФУНКЦИЙ мх ДНК В КЛЕТКЕ С ВЫСОКИМ ПОТРЕБЛЕНИЕМ ЭНЕРГИИ В клетке одно ядро,
- 70. ПРИМЕР Клинические последствия порогового эффекта при митохондриальных болезнях Относительно низкие уровни (10-50 %) мутации не причинят
- 71. 2 РАЗЛИЧИЕ ФЕНОТИПИЧЕСКИХ ПРОЯВЛЕНИЙ ПРИ РАЗЛИЧНОМ КОЛИЧЕСТВЕ МУТАНТНЫХ МХ ДНК КЛЕТКИ ГОМОПЛАЗМИЯ и ГЕТЕРОПЛАЗМИЯ ГОМОПЛАЗМИЯ МХ
- 72. 2 РАЗЛИЧИЕ ФЕНОТИПИЧЕСКИХ ПРОЯВЛЕНИЙ ПРИ РАЗЛИЧНОМ КОЛИЧЕСТВЕ МУТАНТНЫХ мхДНК КЛЕТКИ ГОМОПЛАЗМИЯ и ГЕТЕРОПЛАЗМИЯ Фенотипические последствия наличия
- 73. ПРИМЕР последствия ГЕТЕРОПЛАЗМИИ при митохондриальных болезнях синдром KSS ( Kearns-Sayre Syndrom ) пациенты имеютдо 80 %
- 74. ПРИМЕР последствия ГЕТЕРОПЛАЗМИИ при митохондриальных болезнях ДРАМАТИЧЕСКИЙ ПРИМЕР ТЕРАПИИ У детей, страдающих синдромом Pearson для коррекции
- 75. 3 ИЗОЛЯЦИЯ МИТОТИЧЕСКОЙ АКТИВНОСТИ МХ ДНК ОТ КЛЕТОЧНОГО ЦИКЛА ПОСЛЕДСТВИЯ ЭТОЙ ОСОБЕННОСТИ Если пациент- гетероплазмичен то
- 76. 4 САМОПРОГРЕССИРОВАНИЕ В процессе индивидуального развития распределение клонов мутированной мтДНК в тканях организма человека носит случайный
- 77. Образование свободных радикалов кислорода (АФК) в ДЦ Большинство е– от субстратов, переносится через НАД, КоQ и
- 78. Повреждение клетки свободными радикалами кислорода
- 79. Мутационное воздействие оксидативного стресса на мх ДНК - образование активных форм кислорода (АФК) - оксидативный стресс
- 80. МУТАЦИИ В ГЕНАХ тРНК и их последствия
- 81. Мутации в генах тРНК синдром MERRF ("myoclonus epilepsy with RRF" миоклональная эпилепсия с RRF КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
- 82. Мутации в генах тРНК ПРИЧИНЫ РАЗВИТИЯ MERRF точечная мутация в гене лизиновой тРНК мхДНК приводит к
- 83. Мутации в генах тРНК ПРОЯВЛЕНИЕ MERRF Риск проявления наследования болезни можно предсказать по доле мутантной мхДНК
- 84. Мутации в генах тРНК синдром MELAS ("mitochondrial encephalo myopathy with lactic acidosis and stroke like episodes"
- 85. Мутации в генах тРНК ПРИЧИНЫ РАЗВИТИЯ MELAS точечная мутация в гене ND4 лейциновой тРНК MT TL1*
- 86. МУТАЦИИ В ГЕНАХ рРНК и их последствия
- 87. Мутации в генах рРНК ПРОЯВЛЕНИЯ БОЛЕЗНЕЙ DEAF - нейро сенсорная глухота развитие глухоты в ответ на
- 88. Мутации в генах рРНК DEAF НЕЙРОСЕНСОРНАЯ ГЛУХОТА Мутация в одном из двух закодированных в мхДНК генах
- 89. Мутации в генах рРНК ФЕНОТИПИЧЕСКОЕ ПРОЯВЛЕНИЕ МУТАЦИИ Мутация в гене 16S рРНК приводит к развитию резистентности
- 90. МУТАЦИИ, СНИЖАЮЩИЕ ЧИСЛО КОПИЙ мхДНК и их последствия
- 91. МУТАЦИИ СНИЖАЮЩИЕ ЧИСЛО КОПИЙ мхДНК MILS - летальная инфантильная дыхательная недостаточность MILAS синдром молочнокислого ацидоза миопатия,
- 92. МУТАЦИИ СНИЖАЮЩИЕ ЧИСЛО КОПИЙ мхДНК ПРИЧИНЫ РАЗВИТИЯ Может быть вызвана токсинами окружающей среды ФАКТЫ Лечение СПИДа
- 93. Заключение Классификация митохондриальных болезней человека
- 94. Выделяют 2 группы митохондриальных заболеваний 1) Наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные белки (синдром
- 95. Классификация митохондриальных болезней 1 .Миссенс-мутантные (аминокислотные замены в компонентах I III IV дыхательной цепи) LHON -
- 96. Классификация митохондриальных болезней З. Делеции или дупликации участков мхДНК РЕО - наружная офтальмоплегия KSS - синдром
- 97. Классификация митохондриальных болезней 5. Мутации в генах рРНК DEAF - нейро сенсорная глухота развитие глухоты в
- 98. МЕХАНИЗМ НАСЛЕДОВАНИЯ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ ядерным и митохондриальным локусами
- 99. ДВУХЛОКУСНЫЙ МЕХАНИЗМ НАСЛЕДОВАНИЯ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ Многие митохондриальные белки кодируются генами ядерного генома, синтезируются в цитоплазме, а
- 100. ДВУХЛОКУСНЫЙ МЕХАНИЗМ НАСЛЕДОВАНИЯ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ Существует предположение, что некоторые болезни МХ я МХ могут быть обусловлены
- 101. Существуют ли клинические случаи связанные с мутациями в мхДНК ? 1 КАРДИОПАТИИ различного генеза встречаются в
- 102. Существуют ли связи других сотсояний с мутациями в мхДНК ? Мутационные мхДНК могут играть существенную роль
- 104. Скачать презентацию
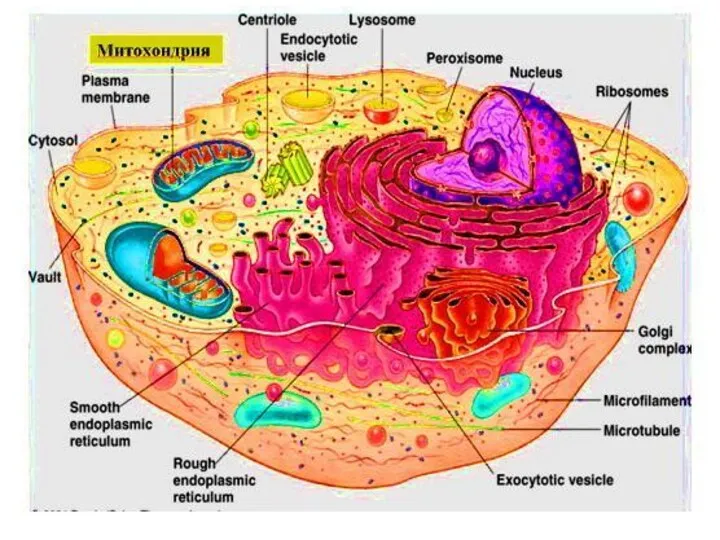
Содержимое клетки
Содержимое клетки


Немного истории
1949 год B.Ephrussi с соавторами
открывает "цитоплазматическую" наследованную
"малую"
Немного истории
1949 год B.Ephrussi с соавторами
открывает "цитоплазматическую" наследованную
"малую"

Немного истории
1962 год - R.Luft c сотрудниками
впервые описал первую болезнь
Немного истории
1962 год - R.Luft c сотрудниками
впервые описал первую болезнь

ЭНДОСИМБИОНТЫ
митохондрии и хлоропласты
(с ними связан процесс
трансдукции энергии внутрь
ЭНДОСИМБИОНТЫ
митохондрии и хлоропласты
(с ними связан процесс
трансдукции энергии внутрь

ЭНДОСИМБИОНТЫ
принесли в клетку бактериальный геном,
остатки которого продолжают
существовать сегодня в виде
ЭНДОСИМБИОНТЫ
принесли в клетку бактериальный геном,
остатки которого продолжают
существовать сегодня в виде

ЭНДОСИМБИОТИЧЕСКАЯ ГИПОТЕЗА
была выдвинута 100 лет назад -
однако факты, подтверждающие эту
теорию были
ЭНДОСИМБИОТИЧЕСКАЯ ГИПОТЕЗА
была выдвинута 100 лет назад -
однако факты, подтверждающие эту
теорию были

Происхождение органелл клетки
Определение происхождения органелл
осуществляют по рРНК
Сравнение гомологичных последовательностей
Происхождение органелл клетки
Определение происхождения органелл
осуществляют по рРНК
Сравнение гомологичных последовательностей


Происхождение органелл клетки
Определение происхождения органелл
осуществляют по рРНК
Сравнение гомологичных последовательностей
Происхождение органелл клетки
Определение происхождения органелл
осуществляют по рРНК
Сравнение гомологичных последовательностей

Происхождение органелл клетки
Гены рРНК расположены среди нескольких генов,
встречающихся как
Происхождение органелл клетки
Гены рРНК расположены среди нескольких генов,
встречающихся как

Происхождение органелл клетки
Хлоропласты и митохондрии пришли из совершенно
различных групп
Происхождение органелл клетки
Хлоропласты и митохондрии пришли из совершенно
различных групп

Строение митохондрий
Общий вид органеллы
Строение митохондрий
Общий вид органеллы
Строение митохондрий
Строение митохондрий
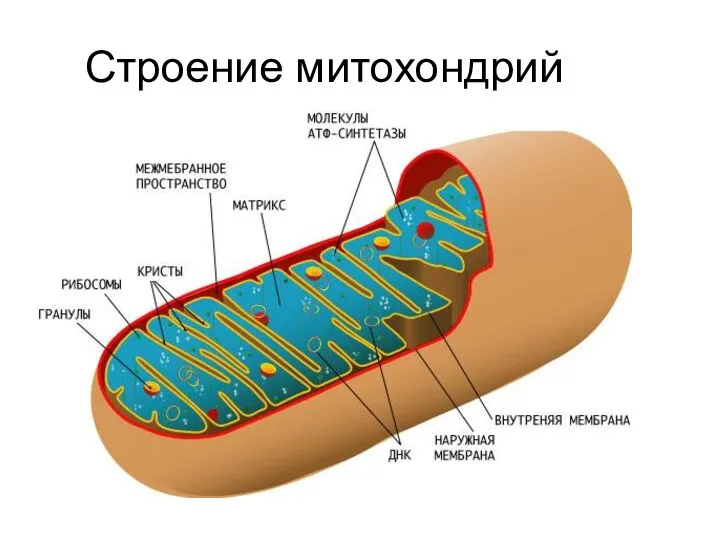
Строение митохондрий
Строение митохондрий

Строение митохондрий
Строение митохондрий

Строение митохондрий
Электронное фото
Строение митохондрий
Электронное фото
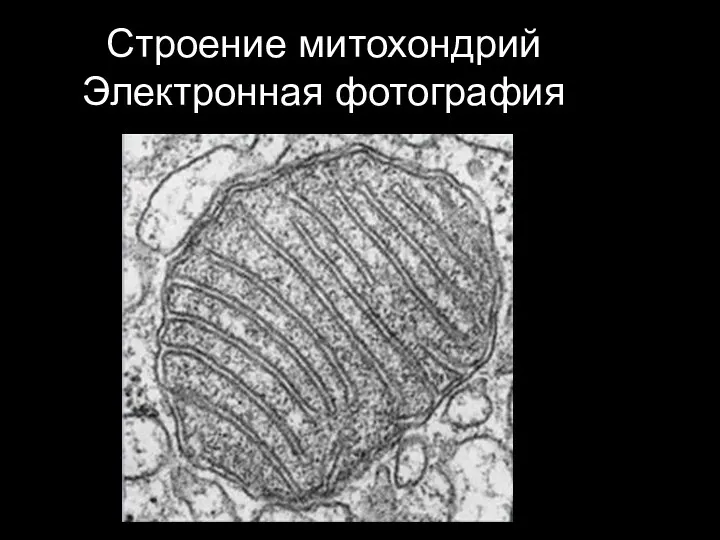
Строение митохондрий
Электронная фотография
Строение митохондрий
Электронная фотография

Питер МИТЧЕЛЛ
Нобелевская премия 1978 года
Питер МИТЧЕЛЛ
Нобелевская премия 1978 года
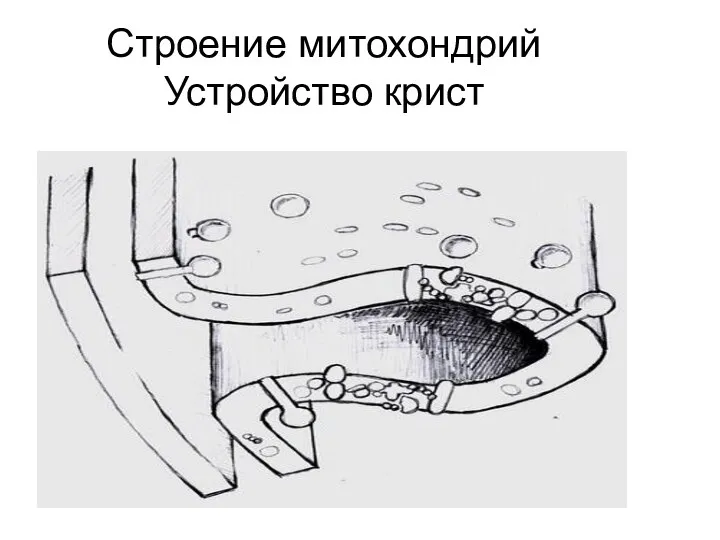
Строение митохондрий
Устройство крист
Строение митохондрий
Устройство крист

Механизм генерации энергии
Механизм генерации энергии
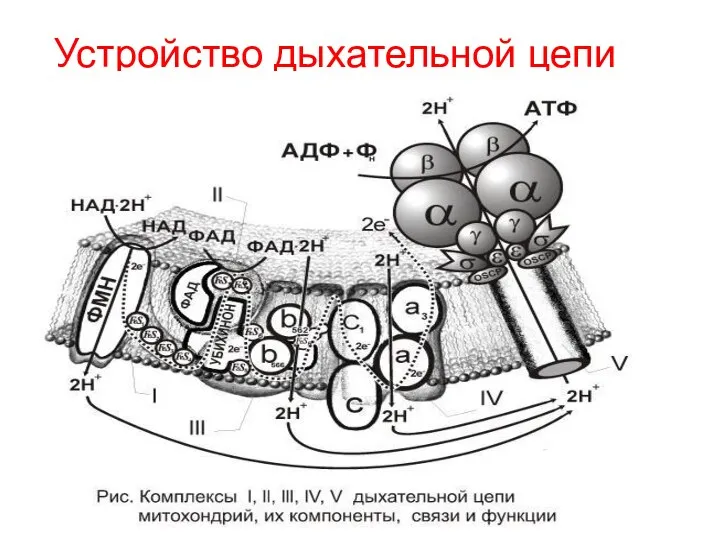
Устройство дыхательной цепи
Устройство дыхательной цепи

Количество мхДНК в клетках
одна митохондрия содержит около 10 молекул мтДНК
Количество мхДНК в клетках
одна митохондрия содержит около 10 молекул мтДНК

В одной клетке от 100 до 1000 митохондрий
В одной клетке от 100 до 1000 митохондрий

Строение ДНК митохондрий
Митохондриальный геном человека был идентифицирован
в 1960 году
Строение ДНК митохондрий
Митохондриальный геном человека был идентифицирован
в 1960 году

Геном митохондриальной ДНК
Геном митохондриальной ДНК


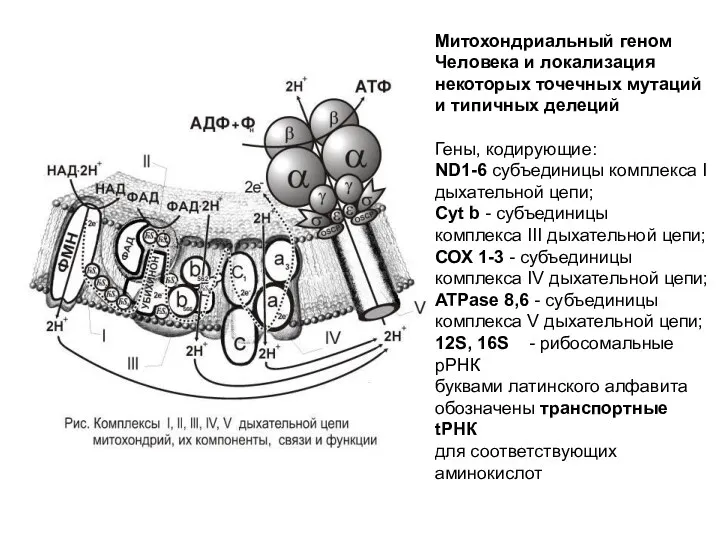
Митохондриальный геном
Человека и локализация некоторых точечных мутаций и типичных делеций
Митохондриальный геном
Человека и локализация некоторых точечных мутаций и типичных делеций

Геном митохондриальной ДНК
16.569 основных нуклеотидных пар
на них закодировано 37
Геном митохондриальной ДНК
16.569 основных нуклеотидных пар
на них закодировано 37

Отличие генома митохондрий
Общий принцип построения геномов митохондрий -
максимальная структурная
Отличие генома митохондрий
Общий принцип построения геномов митохондрий -
максимальная структурная

Отличие генома митохондрий
Генетический код митохондрий позвоночных (человека)
22 антикодона тРНК «узнают»
Отличие генома митохондрий
Генетический код митохондрий позвоночных (человека)
22 антикодона тРНК «узнают»

Отличие генома митохондрий
Экономичность генома достигается
благодаря
1 отсутствию интронов в структурных
Отличие генома митохондрий
Экономичность генома достигается
благодаря
1 отсутствию интронов в структурных

Отличие генома митохондрий
Причина отличия митохондриального кода от ядерного
Для нейтрализации АФК
Отличие генома митохондрий
Причина отличия митохондриального кода от ядерного
Для нейтрализации АФК

Наследование митохондриальных болезней
происходит только по материнской линии
Митохондрии материнской яйцеклетки
содержат
Наследование митохондриальных болезней
происходит только по материнской линии
Митохондрии материнской яйцеклетки
содержат
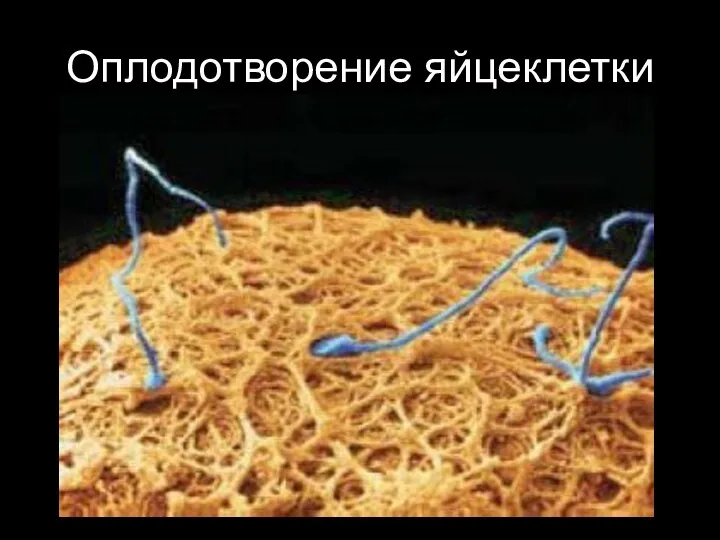
Оплодотворение яйцеклетки
Оплодотворение яйцеклетки

Опять во всем виновата Ева ?
Опять во всем виновата Ева ?

Болезнь первая - синдром Luft
1960 год
Rolf Luft и Lars Ernster
Болезнь первая - синдром Luft
1960 год
Rolf Luft и Lars Ernster

Частота встречаемости болезней митохондрий
в Англии - 1 на 5 000
Частота встречаемости болезней митохондрий
в Англии - 1 на 5 000

Причина митохондриальных болезней
Глубокое вовлечение в процесс дефекта митохондриальной
продукции энергии
Причина митохондриальных болезней
Глубокое вовлечение в процесс дефекта митохондриальной
продукции энергии

Типичные симптомы мх болезней
Известно около 50 митохондриальных болезней.
В их
Типичные симптомы мх болезней
Известно около 50 митохондриальных болезней.
В их

Клинический диагноз
митохондриальной болезни основан
1. Биохимических признаках
(выраженный молочнокислый
Клинический диагноз
митохондриальной болезни основан
1. Биохимических признаках
(выраженный молочнокислый

Подход к классификации митохондриальных болезней
1.
Смысловые замены в структурных
Подход к классификации митохондриальных болезней
1.
Смысловые замены в структурных

ВИДЫ МУТАЦИЙ мхДНК
и вызванные этими нарушениями
митохондриальные болезни
ВИДЫ МУТАЦИЙ мхДНК
и вызванные этими нарушениями
митохондриальные болезни

Точковые мутации
и их последствия
Точковые мутации
и их последствия

Точковые мутации
Болезнь Лебера
Болезнь Лебера
Leber's hereditary optic neuropathy - LHON
наследственная
Точковые мутации
Болезнь Лебера
Болезнь Лебера
Leber's hereditary optic neuropathy - LHON
наследственная

Точковые мутации
Основной исследователь –
Douglas Wallace и сотрудники
(1988)
Точковые мутации
Основной исследователь –
Douglas Wallace и сотрудники
(1988)

Точковые мутации
Болезнь Лебера
ПРИЧИНЫ РАЗВиТИЯ БОЛЕЗНИ
Данная мутация выбивает
один кодон
Точковые мутации
Болезнь Лебера
ПРИЧИНЫ РАЗВиТИЯ БОЛЕЗНИ
Данная мутация выбивает
один кодон

Точковые мутации
Болезнь Лебера
Болезнь чаще поражает мужчин (в соотношении 4: 1,3:
Точковые мутации
Болезнь Лебера
Болезнь чаще поражает мужчин (в соотношении 4: 1,3:

Точковые мутации
Болезнь Лебера
Клинические признаки при данных мутациях
различной локализации существенно не
Точковые мутации
Болезнь Лебера
Клинические признаки при данных мутациях
различной локализации существенно не

Точковые мутации
Болезнь Лебера
Со времени открытия гиганских делеций
и точечных мутаций
Точковые мутации
Болезнь Лебера
Со времени открытия гиганских делеций
и точечных мутаций

Точковые мутации
Пигментный ретинит
Пигментный ретинит NARP
("neuropathy, ataxia, retinitis,
Точковые мутации
Пигментный ретинит
Пигментный ретинит NARP
("neuropathy, ataxia, retinitis,

Точковые мутации
Пигментный ретинит
ПРИЧИНА
Наличие точечной мутации гена
6-ой субъединицы Н+АТФазы,
Точковые мутации
Пигментный ретинит
ПРИЧИНА
Наличие точечной мутации гена
6-ой субъединицы Н+АТФазы,

ДЕЛЕЦИИ мхДНК
(ВЫПАДЕНИЕ ЕНЕТИЧЕСКОЙ ИНФОРМАЦИИ)
и их последствия
ДЕЛЕЦИИ мхДНК
(ВЫПАДЕНИЕ ЕНЕТИЧЕСКОЙ ИНФОРМАЦИИ)
и их последствия

Делеции мхДНК
1 РЕО - "progressive external ophthalmoplegia"
(прогрессирующей экстраофтальмоплегией)
Делеции мхДНК
1 РЕО - "progressive external ophthalmoplegia"
(прогрессирующей экстраофтальмоплегией)

Делеции мхДНК
ОСНОВНОЙ ИССЛЕДОВАТЕЛЬ
Ian Holt и его коллеги идентифицировали
пациентов с
Делеции мхДНК
ОСНОВНОЙ ИССЛЕДОВАТЕЛЬ
Ian Holt и его коллеги идентифицировали
пациентов с

Делеции мхДНК
ПРИЧИНЫ РАЗВИТИЯ БОЛЕЗНИ
Крупные делеции мхДНК обычно БЛОКИРУЮТ
ТРАНСКРИПЦИЮ всех
Делеции мхДНК
ПРИЧИНЫ РАЗВИТИЯ БОЛЕЗНИ
Крупные делеции мхДНК обычно БЛОКИРУЮТ
ТРАНСКРИПЦИЮ всех

Делеции мх ДНК
ОБЩИЕ ПРИЗНАКИ
одиночные проявления или
являющихся частью мультисистемных
Делеции мх ДНК
ОБЩИЕ ПРИЗНАКИ
одиночные проявления или
являющихся частью мультисистемных

Делеции ДНК
PEO (прогрессирующая экстраофтальмоплегия)
Клинически проявляется параличом
экстраокулярных мускул, включая
Делеции ДНК
PEO (прогрессирующая экстраофтальмоплегия)
Клинически проявляется параличом
экстраокулярных мускул, включая

Делеции мхДНК
Синдром Kearns-Sayre
("Kearns-Sayre syndrome" - KSS)
Клинически проявляется в
Делеции мхДНК
Синдром Kearns-Sayre
("Kearns-Sayre syndrome" - KSS)
Клинически проявляется в

Делеции мх ДНК
ОБЩИЕ ПРИЗНАКИ
эти мутации потомством не наследуются, так
Делеции мх ДНК
ОБЩИЕ ПРИЗНАКИ
эти мутации потомством не наследуются, так

Делеции мх ДНК
ОБЩИЕ ПРИЗНАКИ
при незначительном содержании
делетированной мхДНК в
Делеции мх ДНК
ОБЩИЕ ПРИЗНАКИ
при незначительном содержании
делетированной мхДНК в

Делеции мх ДНК
фенотипические проявления одного и того же дефекта делеции
Делеции мх ДНК
фенотипические проявления одного и того же дефекта делеции

ДУПЛИКАЦИЯ мхДНК
и их последствия
ДУПЛИКАЦИЯ мхДНК
и их последствия

Дупликация мх ДНК
ПРИЧИНЫ РАЗВИТИЯ БОЛЕЗНИ
две формы мхДНК – одна
Дупликация мх ДНК
ПРИЧИНЫ РАЗВИТИЯ БОЛЕЗНИ
две формы мхДНК – одна

Дупликация мх ДНК
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
- митохондриальные миопатии
- множественные поражения других
Дупликация мх ДНК
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
- митохондриальные миопатии
- множественные поражения других

ОСОБЕННОСТИ
МИТОХОНДРИАЛЬНЫХ
БОЛЕЗНЕЙ
ОСОБЕННОСТИ
МИТОХОНДРИАЛЬНЫХ
БОЛЕЗНЕЙ

1 НИЗКИЕ ПОРОГИ ДИСФУНКЦИЙ мх ДНК В КЛЕТКЕ С ВЫСОКИМ ПОТРЕБЛЕНИЕМ
1 НИЗКИЕ ПОРОГИ ДИСФУНКЦИЙ мх ДНК В КЛЕТКЕ С ВЫСОКИМ ПОТРЕБЛЕНИЕМ

ПРИМЕР Клинические последствия
порогового эффекта при митохондриальных болезнях
Относительно низкие уровни
ПРИМЕР Клинические последствия
порогового эффекта при митохондриальных болезнях
Относительно низкие уровни

2 РАЗЛИЧИЕ ФЕНОТИПИЧЕСКИХ ПРОЯВЛЕНИЙ ПРИ РАЗЛИЧНОМ КОЛИЧЕСТВЕ МУТАНТНЫХ МХ ДНК КЛЕТКИ
2 РАЗЛИЧИЕ ФЕНОТИПИЧЕСКИХ ПРОЯВЛЕНИЙ ПРИ РАЗЛИЧНОМ КОЛИЧЕСТВЕ МУТАНТНЫХ МХ ДНК КЛЕТКИ

2 РАЗЛИЧИЕ ФЕНОТИПИЧЕСКИХ ПРОЯВЛЕНИЙ ПРИ РАЗЛИЧНОМ КОЛИЧЕСТВЕ МУТАНТНЫХ мхДНК КЛЕТКИ
2 РАЗЛИЧИЕ ФЕНОТИПИЧЕСКИХ ПРОЯВЛЕНИЙ ПРИ РАЗЛИЧНОМ КОЛИЧЕСТВЕ МУТАНТНЫХ мхДНК КЛЕТКИ

ПРИМЕР последствия ГЕТЕРОПЛАЗМИИ
при митохондриальных болезнях
синдром KSS ( Kearns-Sayre Syndrom )
пациенты
ПРИМЕР последствия ГЕТЕРОПЛАЗМИИ
при митохондриальных болезнях
синдром KSS ( Kearns-Sayre Syndrom )
пациенты

ПРИМЕР последствия ГЕТЕРОПЛАЗМИИ
при митохондриальных болезнях
ДРАМАТИЧЕСКИЙ ПРИМЕР ТЕРАПИИ
У детей, страдающих
ПРИМЕР последствия ГЕТЕРОПЛАЗМИИ
при митохондриальных болезнях
ДРАМАТИЧЕСКИЙ ПРИМЕР ТЕРАПИИ
У детей, страдающих

3 ИЗОЛЯЦИЯ МИТОТИЧЕСКОЙ АКТИВНОСТИ МХ ДНК
ОТ КЛЕТОЧНОГО ЦИКЛА
ПОСЛЕДСТВИЯ ЭТОЙ
3 ИЗОЛЯЦИЯ МИТОТИЧЕСКОЙ АКТИВНОСТИ МХ ДНК
ОТ КЛЕТОЧНОГО ЦИКЛА
ПОСЛЕДСТВИЯ ЭТОЙ

4 САМОПРОГРЕССИРОВАНИЕ
В процессе индивидуального развития
распределение клонов мутированной мтДНК в
4 САМОПРОГРЕССИРОВАНИЕ
В процессе индивидуального развития
распределение клонов мутированной мтДНК в

Образование свободных радикалов кислорода (АФК) в ДЦ
Большинство е– от субстратов,
Образование свободных радикалов кислорода (АФК) в ДЦ
Большинство е– от субстратов,
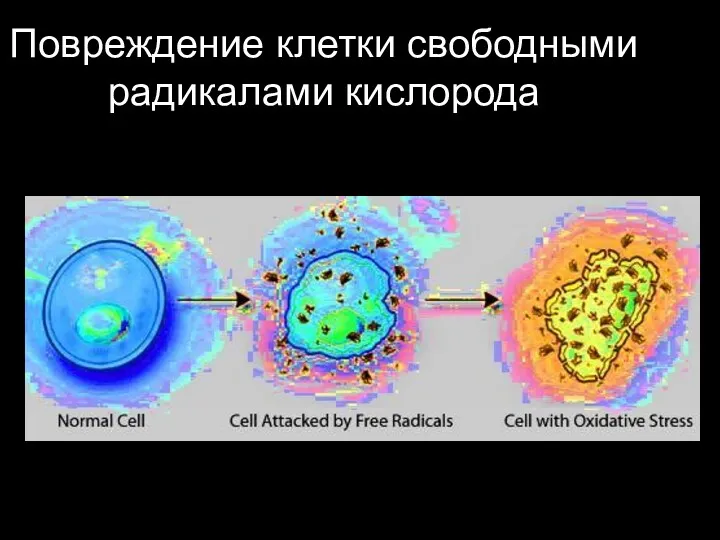
Повреждение клетки свободными радикалами кислорода
Повреждение клетки свободными радикалами кислорода

Мутационное воздействие оксидативного стресса на мх ДНК
- образование активных форм
Мутационное воздействие оксидативного стресса на мх ДНК
- образование активных форм

МУТАЦИИ В ГЕНАХ тРНК
и их последствия
МУТАЦИИ В ГЕНАХ тРНК
и их последствия

Мутации в генах тРНК
синдром MERRF ("myoclonus epilepsy with RRF"
миоклональная эпилепсия
Мутации в генах тРНК
синдром MERRF ("myoclonus epilepsy with RRF"
миоклональная эпилепсия

Мутации в генах тРНК
ПРИЧИНЫ РАЗВИТИЯ MERRF
точечная мутация в гене лизиновой
Мутации в генах тРНК
ПРИЧИНЫ РАЗВИТИЯ MERRF
точечная мутация в гене лизиновой

Мутации в генах тРНК
ПРОЯВЛЕНИЕ MERRF
Риск проявления наследования болезни можно предсказать
Мутации в генах тРНК
ПРОЯВЛЕНИЕ MERRF
Риск проявления наследования болезни можно предсказать

Мутации в генах тРНК
синдром MELAS ("mitochondrial encephalo myopathy with lactic
acidosis
Мутации в генах тРНК
синдром MELAS ("mitochondrial encephalo myopathy with lactic
acidosis

Мутации в генах тРНК
ПРИЧИНЫ РАЗВИТИЯ MELAS
точечная мутация в гене ND4
Мутации в генах тРНК
ПРИЧИНЫ РАЗВИТИЯ MELAS
точечная мутация в гене ND4

МУТАЦИИ В ГЕНАХ рРНК
и их последствия
МУТАЦИИ В ГЕНАХ рРНК
и их последствия

Мутации в генах рРНК
ПРОЯВЛЕНИЯ БОЛЕЗНЕЙ
DEAF - нейро сенсорная глухота
развитие
Мутации в генах рРНК
ПРОЯВЛЕНИЯ БОЛЕЗНЕЙ
DEAF - нейро сенсорная глухота
развитие

Мутации в генах рРНК
DEAF НЕЙРОСЕНСОРНАЯ ГЛУХОТА
Мутация в одном из двух закодированных
Мутации в генах рРНК
DEAF НЕЙРОСЕНСОРНАЯ ГЛУХОТА
Мутация в одном из двух закодированных

Мутации в генах рРНК
ФЕНОТИПИЧЕСКОЕ ПРОЯВЛЕНИЕ МУТАЦИИ
Мутация в гене 16S рРНК
Мутации в генах рРНК
ФЕНОТИПИЧЕСКОЕ ПРОЯВЛЕНИЕ МУТАЦИИ
Мутация в гене 16S рРНК

МУТАЦИИ, СНИЖАЮЩИЕ ЧИСЛО КОПИЙ мхДНК
и их последствия
МУТАЦИИ, СНИЖАЮЩИЕ ЧИСЛО КОПИЙ мхДНК
и их последствия

МУТАЦИИ СНИЖАЮЩИЕ ЧИСЛО КОПИЙ мхДНК
MILS - летальная инфантильная дыхательная
недостаточность
МУТАЦИИ СНИЖАЮЩИЕ ЧИСЛО КОПИЙ мхДНК
MILS - летальная инфантильная дыхательная
недостаточность

МУТАЦИИ СНИЖАЮЩИЕ ЧИСЛО КОПИЙ мхДНК
ПРИЧИНЫ РАЗВИТИЯ
Может быть вызвана токсинами окружающей
МУТАЦИИ СНИЖАЮЩИЕ ЧИСЛО КОПИЙ мхДНК
ПРИЧИНЫ РАЗВИТИЯ
Может быть вызвана токсинами окружающей

Заключение
Классификация
митохондриальных болезней
человека
Заключение
Классификация
митохондриальных болезней
человека

Выделяют 2 группы митохондриальных заболеваний
1) Наследственные синдромы, обусловленные мутациями генов, ответственных
Выделяют 2 группы митохондриальных заболеваний
1) Наследственные синдромы, обусловленные мутациями генов, ответственных

Классификация митохондриальных болезней
1 .Миссенс-мутантные (аминокислотные замены в
компонентах I
Классификация митохондриальных болезней
1 .Миссенс-мутантные (аминокислотные замены в
компонентах I

Классификация митохондриальных болезней
З. Делеции или дупликации участков мхДНК
РЕО - наружная офтальмоплегия
KSS
Классификация митохондриальных болезней
З. Делеции или дупликации участков мхДНК
РЕО - наружная офтальмоплегия
KSS

Классификация митохондриальных болезней
5. Мутации в генах рРНК
DEAF - нейро сенсорная глухота
Классификация митохондриальных болезней
5. Мутации в генах рРНК
DEAF - нейро сенсорная глухота

МЕХАНИЗМ
НАСЛЕДОВАНИЯ
МИТОХОНДРИАЛЬНЫХ
БОЛЕЗНЕЙ
ядерным
и митохондриальным локусами
МЕХАНИЗМ
НАСЛЕДОВАНИЯ
МИТОХОНДРИАЛЬНЫХ
БОЛЕЗНЕЙ
ядерным
и митохондриальным локусами

ДВУХЛОКУСНЫЙ МЕХАНИЗМ НАСЛЕДОВАНИЯ
МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Многие митохондриальные белки
кодируются генами ядерного генома,
ДВУХЛОКУСНЫЙ МЕХАНИЗМ НАСЛЕДОВАНИЯ
МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Многие митохондриальные белки
кодируются генами ядерного генома,

ДВУХЛОКУСНЫЙ МЕХАНИЗМ НАСЛЕДОВАНИЯ
МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Существует предположение, что некоторые болезни МХ я
ДВУХЛОКУСНЫЙ МЕХАНИЗМ НАСЛЕДОВАНИЯ
МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Существует предположение, что некоторые болезни МХ я

Существуют ли клинические случаи
связанные с мутациями в мхДНК ?
1
Существуют ли клинические случаи
связанные с мутациями в мхДНК ?
1

Существуют ли связи других сотсояний
с мутациями в мхДНК ?
Мутационные мхДНК
Существуют ли связи других сотсояний
с мутациями в мхДНК ?
Мутационные мхДНК
 Асептика и антисептика в процедурном кабинете
Асептика и антисептика в процедурном кабинете Лекарственная болезнь
Лекарственная болезнь Эпидемиология и профилактика холеры
Эпидемиология и профилактика холеры Помощь больным с инсультом
Помощь больным с инсультом Обеспечение помехозащищенности медицинских комплексов
Обеспечение помехозащищенности медицинских комплексов Лейшманиозы
Лейшманиозы Санитарно-противоэпидемические (профилактические) мероприятия при ликвидации последствий чрезвычайных ситуаций
Санитарно-противоэпидемические (профилактические) мероприятия при ликвидации последствий чрезвычайных ситуаций Патофизиология сердечно-сосудистой системы. Лекция 2
Патофизиология сердечно-сосудистой системы. Лекция 2 Дифференциальный диагноз при синдроме бронхиальной обструкции
Дифференциальный диагноз при синдроме бронхиальной обструкции Психотические расстройства вызванные употреблением ПАВ
Психотические расстройства вызванные употреблением ПАВ Federal State Educational Institution of Higher Education
Federal State Educational Institution of Higher Education Суппозитории. Основы для суппозиториев
Суппозитории. Основы для суппозиториев Патоморфологические аспекты антенатальной и перинатальной патологии
Патоморфологические аспекты антенатальной и перинатальной патологии Дерматомиозит
Дерматомиозит Уход за урологическими больными
Уход за урологическими больными Туберкулезге қарсы препараттар, фармакокинетикасы, фармакодинамикасы, жанама әсерлері және оларды жою
Туберкулезге қарсы препараттар, фармакокинетикасы, фармакодинамикасы, жанама әсерлері және оларды жою Лимфогранулематоз. Лимфопролиферативное заболевание
Лимфогранулематоз. Лимфопролиферативное заболевание Респираторный дистресс-синдром взрослых
Респираторный дистресс-синдром взрослых Жансыздандыру кезіндегі жергілікті асқынулар
Жансыздандыру кезіндегі жергілікті асқынулар Анатомо-физиологические особенности девочек различных возрастных периодов, с позиции анестезиолога-реаниматолога
Анатомо-физиологические особенности девочек различных возрастных периодов, с позиции анестезиолога-реаниматолога Глобальные рекомендации по физической активности для здоровья
Глобальные рекомендации по физической активности для здоровья Биполярное аффективное расстройство (F31)
Биполярное аффективное расстройство (F31) Первая помощь при ранении. Повязки
Первая помощь при ранении. Повязки Общие основы лечебной физической культуры
Общие основы лечебной физической культуры Пульмонология. Болезни органов дыхания
Пульмонология. Болезни органов дыхания Көкжөтел
Көкжөтел Органы чувств и анализаторы
Органы чувств и анализаторы Повреждения органов брюшной полости
Повреждения органов брюшной полости