- Моногенные болезни

Содержание
- 2. Моногенные болезни (МБ) — это заболевания, в основе которых лежит мутация одного гена, в результате чего
- 3. В настоящее время известно более 6000 нозологических единиц МБ. На 1000 новорожденных МБ выявляются у 42
- 4. Ген – участок молекулы ДНК, выполняющий определенную функцию Геном – совокупность всех генов организма (всей ДНК
- 5. НУКЛЕОТИД ДНК
- 6. А а Аллельные гены – гены, расположенные в гомологичных локусах гомологичных хромосом и отвечающие за развитие
- 7. Классификация МБ I. По частоте встречаемости: 1. Часто встречающиеся 1 : 10 тыс. новорожденных и чаще
- 8. II. Основная патогенетическая классификация НБО (НДО) – наследственные болезни обмена (ферментопатии) НБО аминокислот (фенилкетонурия) НБО углеводов
- 9. Дефект гена определяет дефект белка-фермента в результате блокируется б/х реакция, количество субстрата в клетке увеличивается, количество
- 10. ФЕНИЛКЕТОНУРИЯ (ФКУ) ФЕНИЛАЛАНИН ТИРОЗИН фенилаланингидроксилаза ГЕН фенилпировиноградная кислота фенилмолочная кислота фенилуксусная кислота
- 11. ФКУ
- 12. Болезнь манифестирует в возрасте 2-6 месяцев. Характерно: вялость ребенка, отсутствие интереса к окружающему; иногда повышенная раздражительность,
- 13. 2. Моногенные синдромы ВПР - синдромы врожденных пороков, развитие которых обусловлено мутацией одного гена
- 14. Синдром МАРФАНА – наследственное заболевание, связанное с нарушениями обмена соединительной ткани. Болезнь описана В. Марфаном в
- 15. С-м Марфана Нарушение опорно- двигательного аппарата Нарушение органа зрения - подвывих хрусталика Изменение сердечно- сосудистой системы-
- 16. - астенический тип телосложения; - дефицит массы тела; - долихостеномелия; - арахнодактилия; - искривление позвоночника; -
- 17. С-м Марфана у девочки 14 лет
- 19. С-м Марфана арахнодактилия
- 20. С-м Марфана арахнодактилия
- 21. Долихостеномелия. Арахнодактилия. Положительный симптом запястья.
- 22. Симптом «большого пальца» при арахнодактилии.
- 23. С-мы большого пальца и запястья
- 24. Марфана с-м – подвывих хрусталика
- 25. IQ - N
- 26. III. По типу мутации: Миссенс – замена одного нуклеотида на другой. Нонсенс – замена нуклеотидов, в
- 27. С-М МАРТИН-БЭЛЛ- СИНДРОМ УМСТВЕННОЙ ОТСТАЛОСТИ С ЛОМКОЙ Х ХРОМОСОМОЙ Ген FMR1 локализован в Х хромосоме В
- 28. Характерный фенотип: - прямоугольное лицо; - большие оттопыренные ушные раковины; выступающий лоб; массивный подбородок; макроорхизм; умственная
- 29. IV. По типу наследования: Аутосомно-доминантный Аутосомно-рецессивный Х-сцепленный доминантный Х-сцепленный рецессивный Y-сцепленный Митохондриальный
- 30. Аутосомно-доминантный тип наследования
- 31. Аутосомно-доминантным называется заболевание, развитие которого обусловлено доминантным геном, который локализован в аутосоме Больной имеет генотип АА
- 32. Характерны два типа родословных: - при заболеваниях, при которых больной доживает до репродуктивного возраста, вступает в
- 33. Вертикальный тип наследования Поражение лиц обоих полов
- 34. Единичный случай рождения больного ребенка в здоровой семье – следствие генеративной генной доминантной мутации Последующий риск
- 35. При неполной пенетрантности гена – в родословной имеются «проскакивающие» поколения
- 36. Аутосомно-рецессивный тип наследования
- 37. Аутосомно-рецессивным называется заболевание, развитие которого обусловлено рецессивным геном, локализованным в аутосоме. Генотип пациента - аа
- 38. Горизонтальный тип наследования Поражение лиц обоих полов Последующий риск – 25%
- 39. «Выщеплению» гомозигот способствует инбридинг - кровнородственный брак
- 40. Сцепленными с полом называются заболевания, гены которых расположены в негомологичных участках половых хромосом Х Y
- 41. Х-сцепленный рецессивный тип наследования (заболевание вызывается рецессивным ген, локализованным в негомологичном участке Х хромосомы)
- 42. Поражение лиц мужского пола Матери – носительницы патологического гена (Х*) ½ ¼
- 43. Особенности клиники МБ I. Широкий клинический полиморфизм, генетическими причинами которого являются: - полиаллелизм; - полилокусность; -
- 44. ПОЛИАЛЛЕЛИЗМ (множественный аллелизм) явление, при котором в генофонде популяции существует более двух аллелей Например, при ФКУ:
- 45. ПОЛИЛОКУСНОСТЬ явление, при котором за синтез белковой молекулы отвечает 2 и более генов. ГЕН 1 ГЕН
- 46. В настоящее время различают: Классическую ФКУ (I типа) - ген 12q22-q24.2 Атипичные формы (ФКУ I –
- 47. различная комбинация генов-модификаторов различная доза патологического гена (АА или Аа) явление геномного импринтинга – различная активность
- 48. ГЕНОМНЫЙ ИМПРИНТИНГ – с-м Прадера-Вилли Причина заболевания – инактивация генов q11-13 хромосомы 15 отцовского происхождения -
- 49. ГЕНОМНЫЙ ИМПРИНТИНГ – с-м Ангельмана Причина заболевания инактивация генов хромосомы 15 материнского происхождения олигофрения приступы судорог,
- 50. 2. Варьирующий возраст начала заболевания Внутриутробно реализуется 25% генных мутаций; До начала пубертата – 45%; В
- 51. 3. Неодновременность проявления признаков Например, при синдроме Марфана: при рождении диагностируется арахнодактилия к 3 годам –
- 52. 4. Наличие у больного редко встречающихся специфических симптомов. Например, вертикальные насечки на мочке уха при синдроме
- 53. С-м Беквита-Видемана Макросомия Макроглоссия Пупочная грыжа Насечки на мочке уха
- 54. МУКОВИСЦИДОЗ Муковисцидоз или кистофиброз — это патология экзокринных желез (бронхиальных, потовых, слезных, слюнных), а также поджелудочной
- 55. Ген болезни локализован в 7q31.1-32 и кодирует белок-регулятор трансмембранной проводимости для ионов хлора (CFTR — кистофиброзный
- 56. Основной патогенетический механизм болезни – увеличение вязкости секрета, выделяемого слизеобразующими железами бронхов, кишечника, поджелудочной железы, семенников
- 57. Меконеальный илеус Кишечная непроходимость у новорожденного Рентгенологическое обследование новорожденного с мекониальной непроходимостью.
- 58. Легочная форма Закупорка просвета мелких респираторных путей Присоединение вторичной инфекции Хронический воспалительный процесс в бронхо-легочной системе:
- 59. Бронхи человека, больного муковисцидозом. Гиперпродукция слизи приводит к тому, что заполненные ею бронхи затрудняют дыхание и
- 60. Кишечная форма МВ Изменение водно-электролитного состава панкреатического сока, его сгущение и затруднение выделения в просвет кишечника
- 61. Клиническая картина кишечной формы муковисцидоза обусловлена недостаточностью ферментативной активности желудочно-кишечного тракта, которая особенно ярко проявляется после
- 62. Смешанная форма МВ
- 63. Неонатальный скрининг – обследование всех новорожденных с целью раннего (доклинического) выявления МБ Показания для проведения скрининга:
- 64. Неонатальный скрининг Забор крови на 5 – 7 сутки жизни
- 65. ФЕНИЛКЕТОНУРИЯ ВРОЖДЕННЫЙ ГИПОТИРЕОЗ ГАЛАКТОЗЕМИЯ АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ МУКОВИЗЦИДОЗ
- 67. Скачать презентацию

Моногенные болезни (МБ) — это заболевания, в основе которых лежит мутация
Моногенные болезни (МБ) — это заболевания, в основе которых лежит мутация

В настоящее время известно более 6000 нозологических единиц МБ.
На 1000
В настоящее время известно более 6000 нозологических единиц МБ.
На 1000

Ген – участок молекулы ДНК, выполняющий определенную функцию
Геном – совокупность всех
Ген – участок молекулы ДНК, выполняющий определенную функцию
Геном – совокупность всех
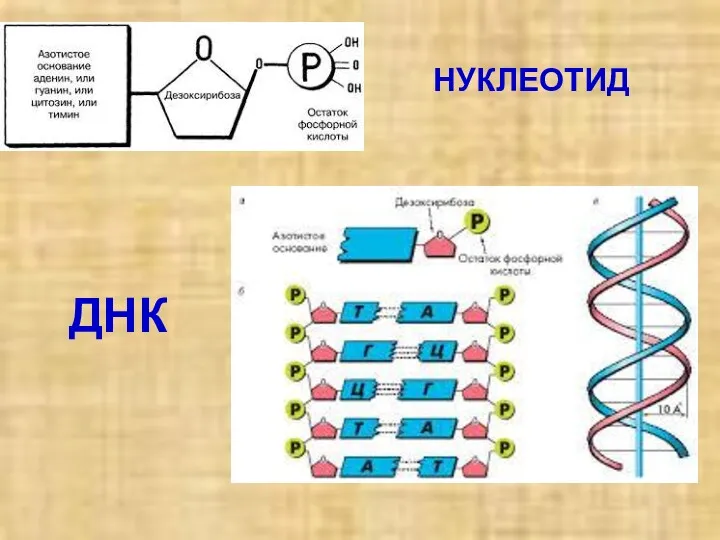
НУКЛЕОТИД
ДНК
НУКЛЕОТИД
ДНК

А
а
Аллельные гены –
гены, расположенные в гомологичных локусах гомологичных хромосом и
А
а
Аллельные гены –
гены, расположенные в гомологичных локусах гомологичных хромосом и

Классификация МБ
I. По частоте встречаемости:
1. Часто встречающиеся
1 : 10 тыс.
Классификация МБ
I. По частоте встречаемости:
1. Часто встречающиеся
1 : 10 тыс.

II. Основная патогенетическая классификация
НБО (НДО) – наследственные болезни обмена (ферментопатии)
НБО аминокислот
II. Основная патогенетическая классификация
НБО (НДО) – наследственные болезни обмена (ферментопатии)
НБО аминокислот
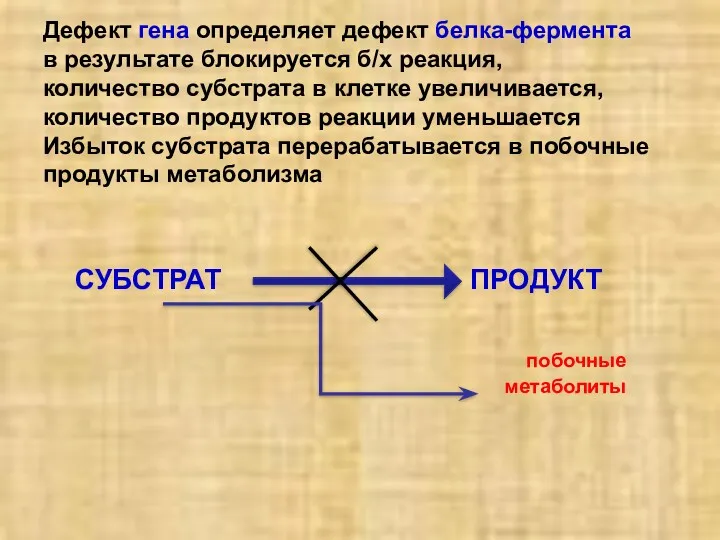
Дефект гена определяет дефект белка-фермента
в результате блокируется б/х реакция,
количество субстрата
Дефект гена определяет дефект белка-фермента в результате блокируется б/х реакция, количество субстрата
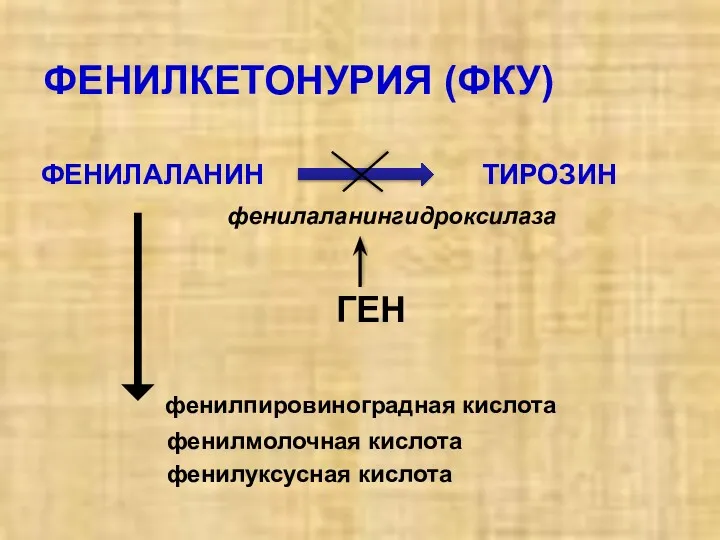
ФЕНИЛКЕТОНУРИЯ (ФКУ)
ФЕНИЛАЛАНИН ТИРОЗИН
фенилаланингидроксилаза
ГЕН
фенилпировиноградная кислота
фенилмолочная кислота
фенилуксусная кислота
ФЕНИЛКЕТОНУРИЯ (ФКУ)
ФЕНИЛАЛАНИН ТИРОЗИН
фенилаланингидроксилаза
ГЕН
фенилпировиноградная кислота
фенилмолочная кислота
фенилуксусная кислота

ФКУ
ФКУ

Болезнь манифестирует в возрасте 2-6 месяцев.
Характерно:
вялость ребенка, отсутствие интереса
Болезнь манифестирует в возрасте 2-6 месяцев.
Характерно:
вялость ребенка, отсутствие интереса

2. Моногенные
синдромы ВПР - синдромы врожденных пороков,
развитие которых
2. Моногенные
синдромы ВПР - синдромы врожденных пороков,
развитие которых

Синдром МАРФАНА –
наследственное заболевание, связанное с нарушениями обмена соединительной ткани.
Синдром МАРФАНА –
наследственное заболевание, связанное с нарушениями обмена соединительной ткани.

С-м Марфана
Нарушение
опорно- двигательного аппарата
Нарушение органа зрения -
подвывих хрусталика
Изменение сердечно-
С-м Марфана
Нарушение
опорно- двигательного аппарата
Нарушение органа зрения -
подвывих хрусталика
Изменение сердечно-
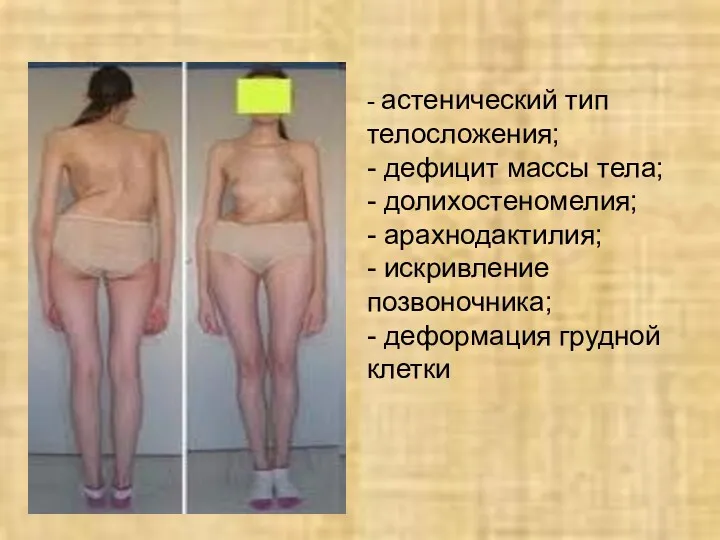
- астенический тип телосложения;
- дефицит массы тела;
- долихостеномелия;
- арахнодактилия;
- искривление позвоночника;
-
- астенический тип телосложения; - дефицит массы тела; - долихостеномелия; - арахнодактилия; - искривление позвоночника; -

С-м Марфана
у девочки
14 лет
С-м Марфана
у девочки
14 лет
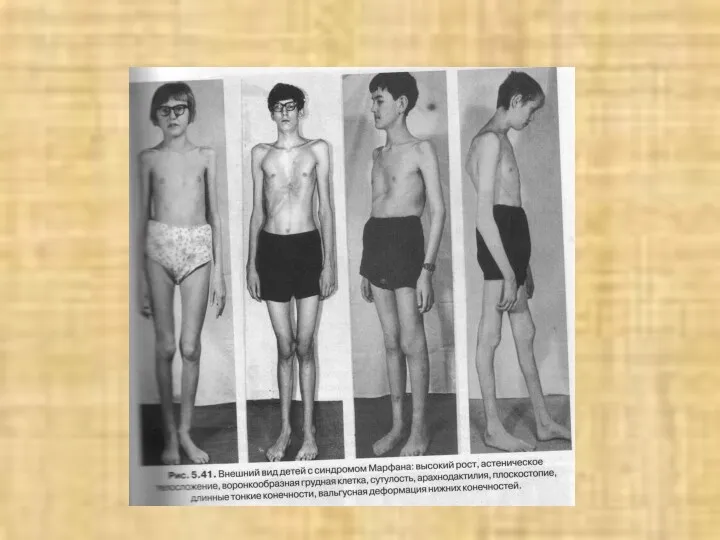

С-м Марфана
арахнодактилия
С-м Марфана
арахнодактилия
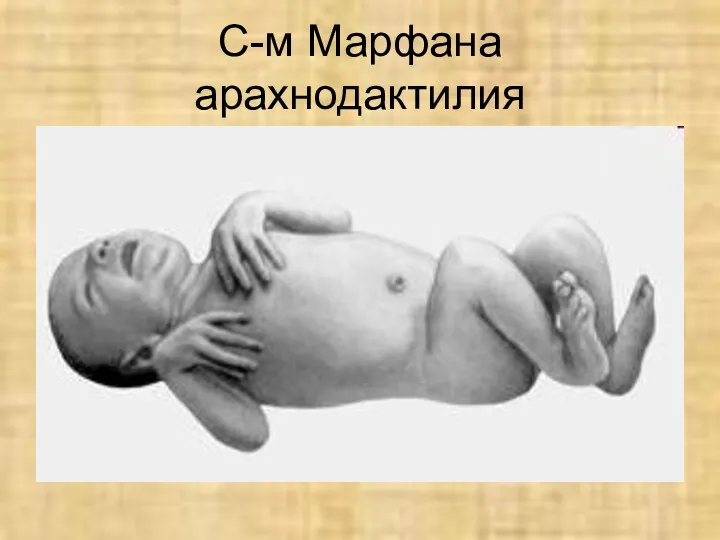
С-м Марфана
арахнодактилия
С-м Марфана
арахнодактилия
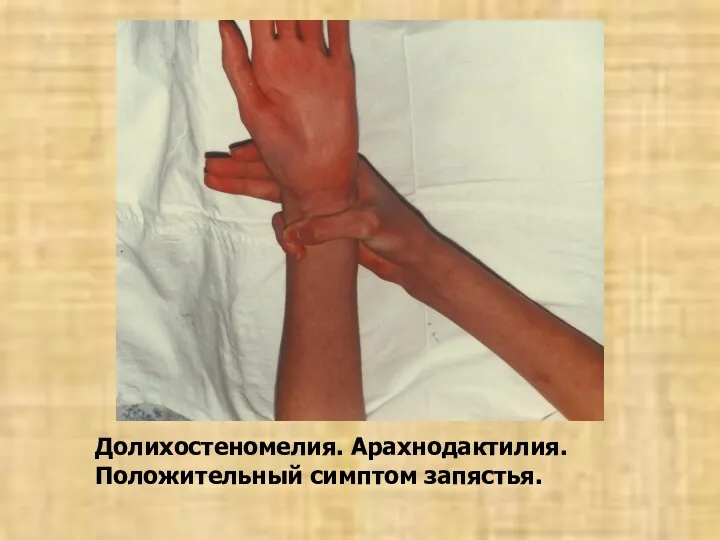
Долихостеномелия. Арахнодактилия.
Положительный симптом запястья.
Долихостеномелия. Арахнодактилия.
Положительный симптом запястья.
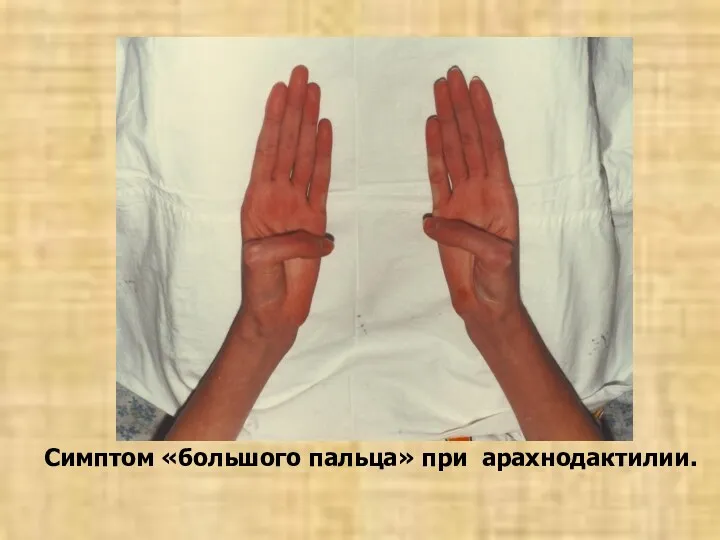
Симптом «большого пальца» при арахнодактилии.
Симптом «большого пальца» при арахнодактилии.
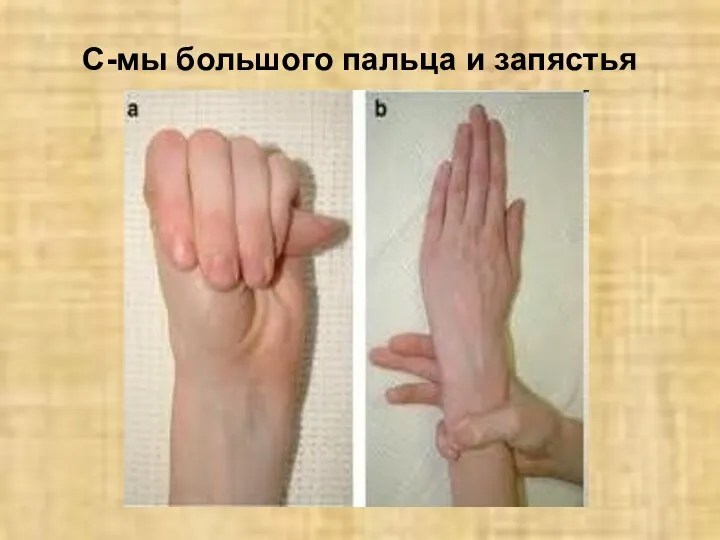
С-мы большого пальца и запястья
С-мы большого пальца и запястья
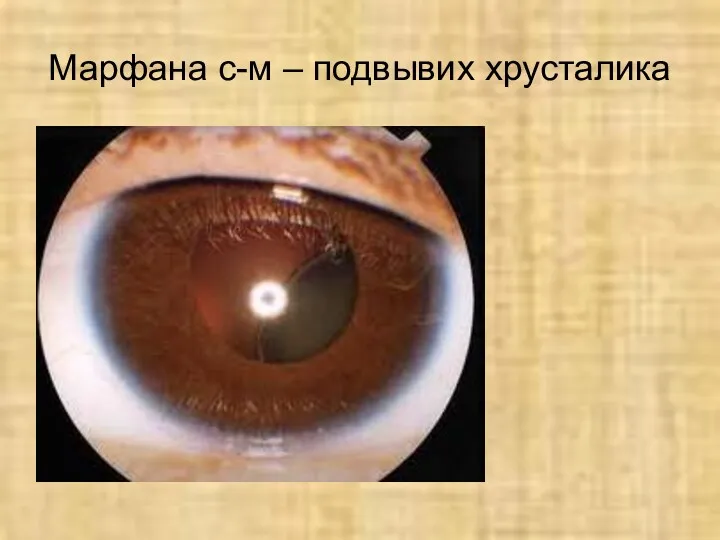
Марфана с-м – подвывих хрусталика
Марфана с-м – подвывих хрусталика

IQ - N
IQ - N

III. По типу мутации:
Миссенс – замена одного нуклеотида на другой.
Нонсенс –
III. По типу мутации: Миссенс – замена одного нуклеотида на другой. Нонсенс –
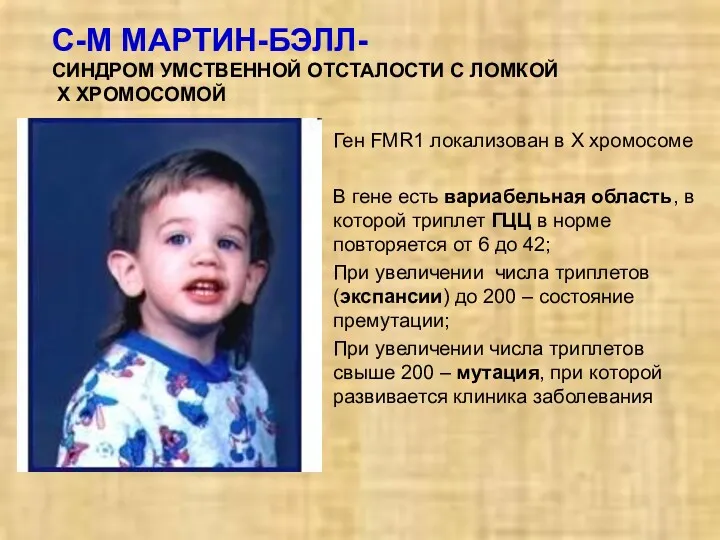
С-М МАРТИН-БЭЛЛ-
СИНДРОМ УМСТВЕННОЙ ОТСТАЛОСТИ С ЛОМКОЙ
Х ХРОМОСОМОЙ
Ген FMR1 локализован
С-М МАРТИН-БЭЛЛ-
СИНДРОМ УМСТВЕННОЙ ОТСТАЛОСТИ С ЛОМКОЙ
Х ХРОМОСОМОЙ
Ген FMR1 локализован

Характерный фенотип:
- прямоугольное лицо;
- большие оттопыренные ушные раковины;
выступающий лоб;
массивный
Характерный фенотип:
- прямоугольное лицо;
- большие оттопыренные ушные раковины;
выступающий лоб;
массивный

IV. По типу наследования:
Аутосомно-доминантный
Аутосомно-рецессивный
Х-сцепленный доминантный
Х-сцепленный рецессивный
Y-сцепленный
Митохондриальный
IV. По типу наследования:
Аутосомно-доминантный
Аутосомно-рецессивный
Х-сцепленный доминантный
Х-сцепленный рецессивный
Y-сцепленный
Митохондриальный

Аутосомно-доминантный тип наследования
Аутосомно-доминантный тип наследования

Аутосомно-доминантным называется заболевание, развитие которого обусловлено доминантным геном, который локализован в
Аутосомно-доминантным называется заболевание, развитие которого обусловлено доминантным геном, который локализован в

Характерны два типа родословных:
- при заболеваниях, при которых больной доживает до
Характерны два типа родословных: - при заболеваниях, при которых больной доживает до
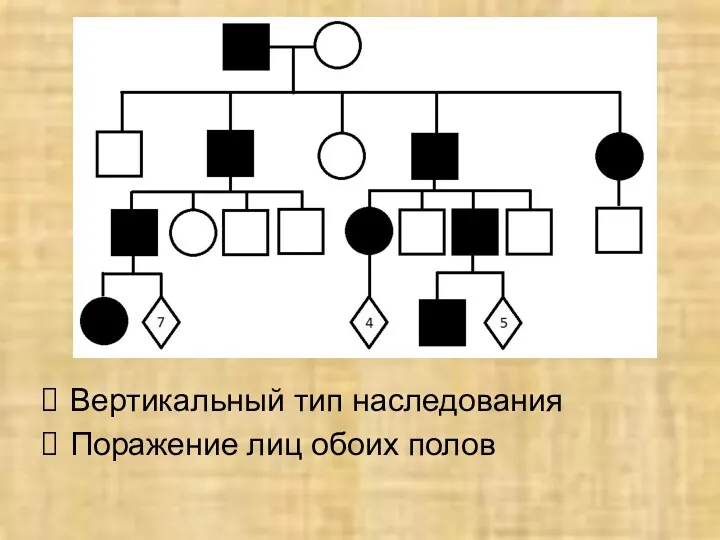
Вертикальный тип наследования
Поражение лиц обоих полов
Вертикальный тип наследования
Поражение лиц обоих полов
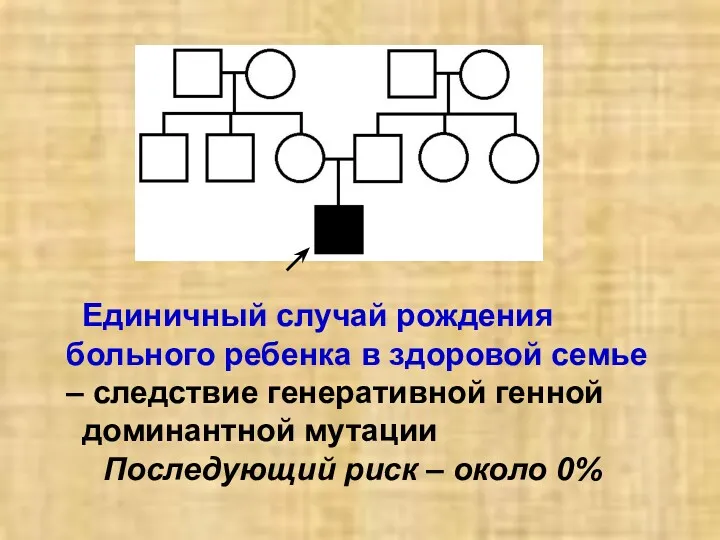
Единичный случай рождения больного ребенка в здоровой семье – следствие генеративной
Единичный случай рождения больного ребенка в здоровой семье – следствие генеративной
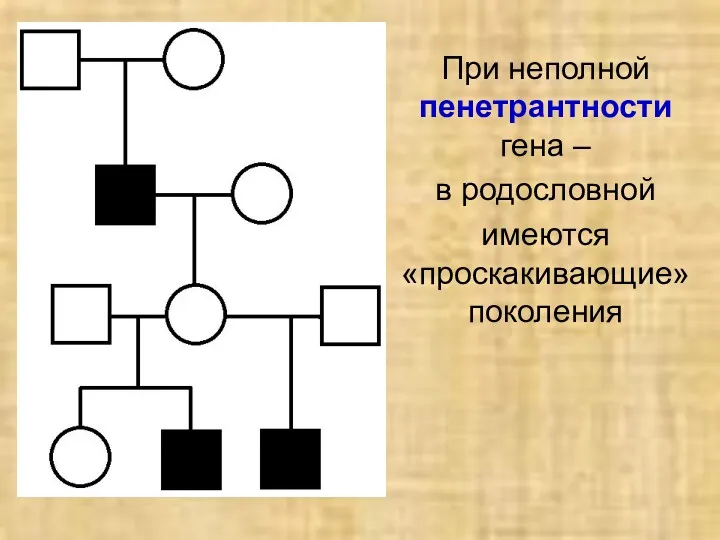
При неполной пенетрантности гена –
в родословной
имеются «проскакивающие» поколения
При неполной пенетрантности гена –
в родословной
имеются «проскакивающие» поколения

Аутосомно-рецессивный тип наследования
Аутосомно-рецессивный тип наследования

Аутосомно-рецессивным называется заболевание, развитие которого обусловлено рецессивным геном, локализованным в аутосоме.
Генотип
Аутосомно-рецессивным называется заболевание, развитие которого обусловлено рецессивным геном, локализованным в аутосоме. Генотип
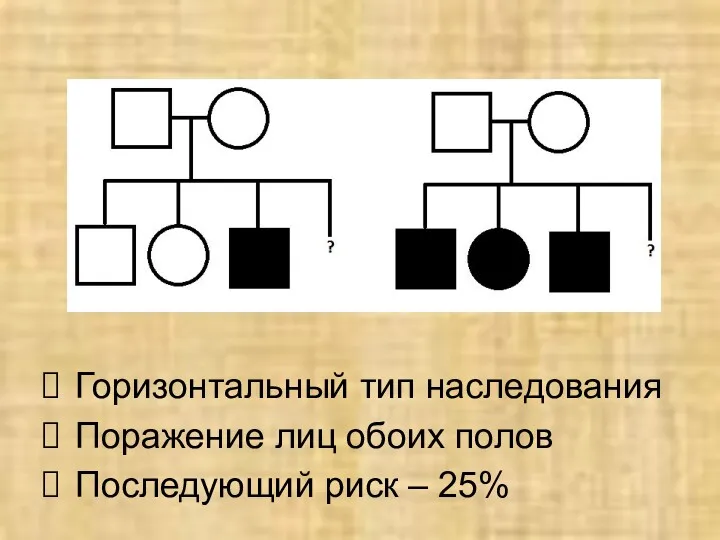
Горизонтальный тип наследования
Поражение лиц обоих полов
Последующий риск – 25%
Горизонтальный тип наследования
Поражение лиц обоих полов
Последующий риск – 25%
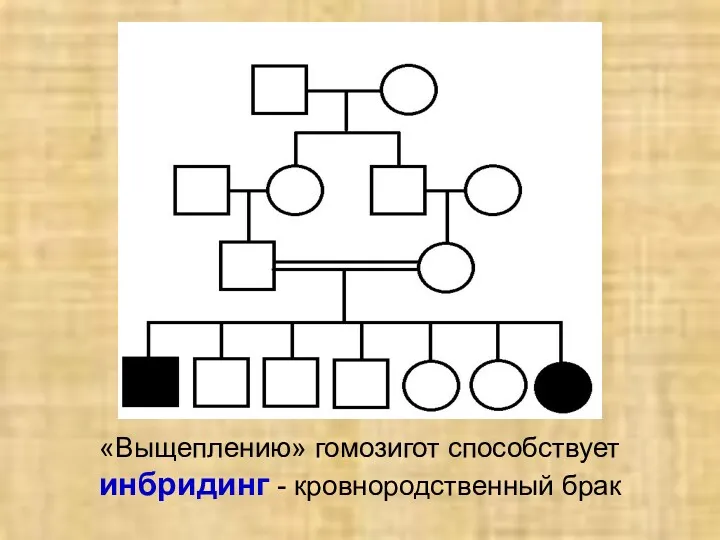
«Выщеплению» гомозигот способствует инбридинг - кровнородственный брак
«Выщеплению» гомозигот способствует инбридинг - кровнородственный брак
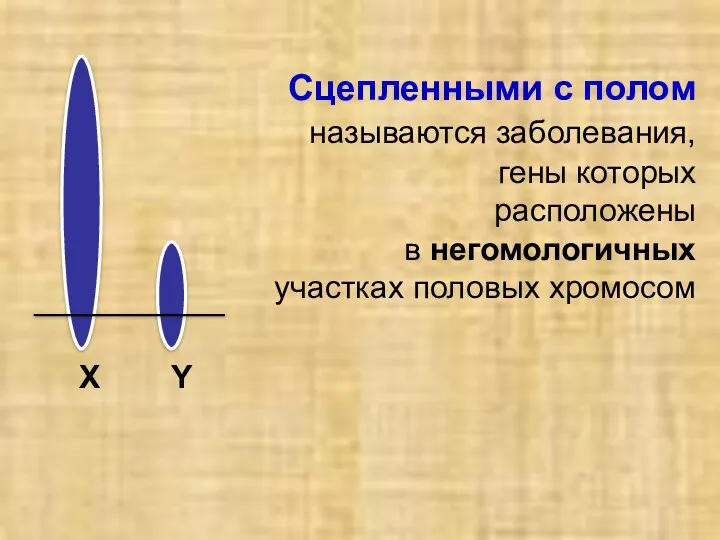
Сцепленными с полом
называются заболевания,
гены которых
расположены
в негомологичных
Сцепленными с полом называются заболевания, гены которых расположены в негомологичных

Х-сцепленный рецессивный тип наследования
(заболевание вызывается рецессивным ген, локализованным в негомологичном участке
Х-сцепленный рецессивный тип наследования (заболевание вызывается рецессивным ген, локализованным в негомологичном участке
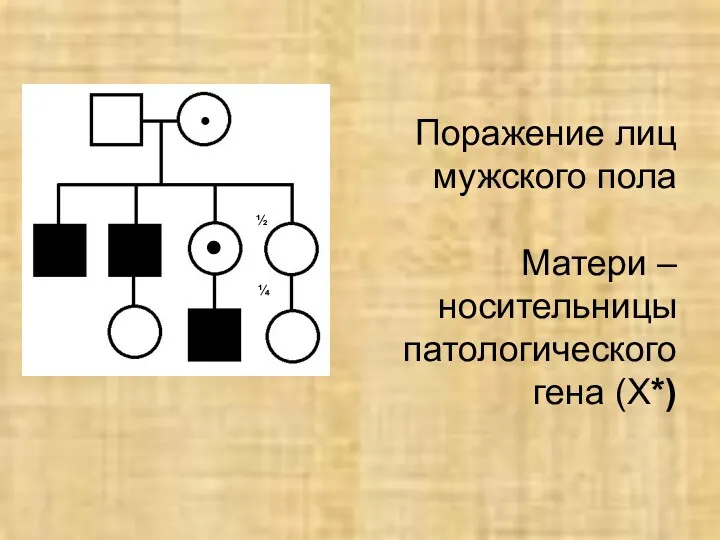
Поражение лиц мужского пола
Матери – носительницы патологического гена (Х*)
½
¼
Поражение лиц мужского пола
Матери – носительницы патологического гена (Х*)
½
¼

Особенности клиники МБ
I. Широкий клинический полиморфизм, генетическими причинами которого являются:
- полиаллелизм;
-
Особенности клиники МБ
I. Широкий клинический полиморфизм, генетическими причинами которого являются:
- полиаллелизм;
-

ПОЛИАЛЛЕЛИЗМ
(множественный аллелизм)
явление, при котором в генофонде популяции существует более двух
ПОЛИАЛЛЕЛИЗМ
(множественный аллелизм)
явление, при котором в генофонде популяции существует более двух
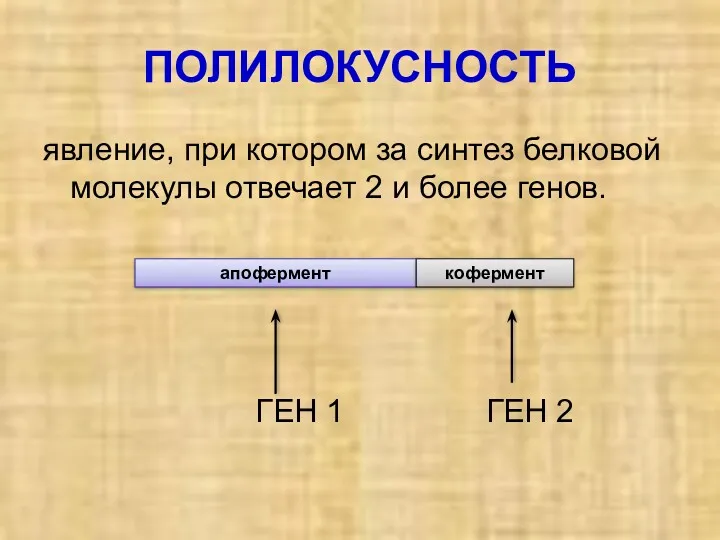
ПОЛИЛОКУСНОСТЬ
явление, при котором за синтез белковой молекулы отвечает 2 и
ПОЛИЛОКУСНОСТЬ
явление, при котором за синтез белковой молекулы отвечает 2 и

В настоящее время различают:
Классическую ФКУ (I типа) - ген 12q22-q24.2
Атипичные формы
В настоящее время различают:
Классическую ФКУ (I типа) - ген 12q22-q24.2
Атипичные формы

различная комбинация генов-модификаторов
различная доза патологического гена (АА или Аа)
явление геномного импринтинга
различная комбинация генов-модификаторов
различная доза патологического гена (АА или Аа)
явление геномного импринтинга
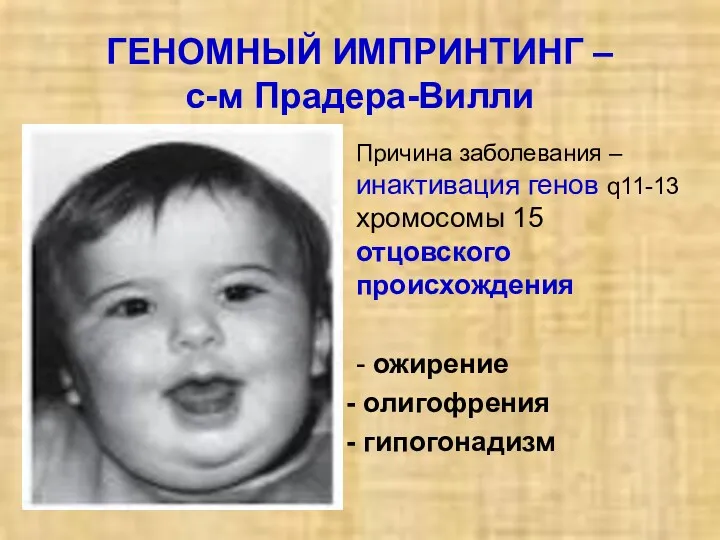
ГЕНОМНЫЙ ИМПРИНТИНГ –
с-м Прадера-Вилли
Причина заболевания – инактивация генов q11-13 хромосомы 15
ГЕНОМНЫЙ ИМПРИНТИНГ –
с-м Прадера-Вилли
Причина заболевания – инактивация генов q11-13 хромосомы 15

ГЕНОМНЫЙ ИМПРИНТИНГ –
с-м Ангельмана
Причина заболевания
инактивация генов
хромосомы 15
материнского происхождения
олигофрения
приступы судорог, резкие
ГЕНОМНЫЙ ИМПРИНТИНГ –
с-м Ангельмана
Причина заболевания
инактивация генов
хромосомы 15
материнского происхождения
олигофрения
приступы судорог, резкие

2. Варьирующий возраст начала заболевания
Внутриутробно реализуется 25% генных мутаций;
До начала пубертата
2. Варьирующий возраст начала заболевания
Внутриутробно реализуется 25% генных мутаций;
До начала пубертата

3. Неодновременность проявления признаков
Например, при синдроме Марфана:
при рождении диагностируется арахнодактилия
к
3. Неодновременность проявления признаков
Например, при синдроме Марфана:
при рождении диагностируется арахнодактилия
к

4. Наличие у больного редко встречающихся специфических симптомов.
Например,
вертикальные насечки
4. Наличие у больного редко встречающихся специфических симптомов.
Например,
вертикальные насечки
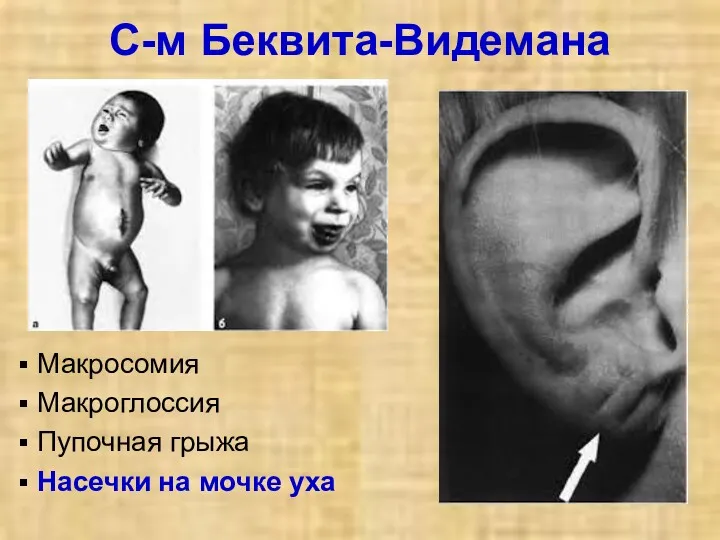
С-м Беквита-Видемана
Макросомия
Макроглоссия
Пупочная грыжа
Насечки на мочке уха
С-м Беквита-Видемана
Макросомия
Макроглоссия
Пупочная грыжа
Насечки на мочке уха

МУКОВИСЦИДОЗ
Муковисцидоз или кистофиброз — это патология экзокринных желез (бронхиальных, потовых, слезных,
МУКОВИСЦИДОЗ
Муковисцидоз или кистофиброз — это патология экзокринных желез (бронхиальных, потовых, слезных,
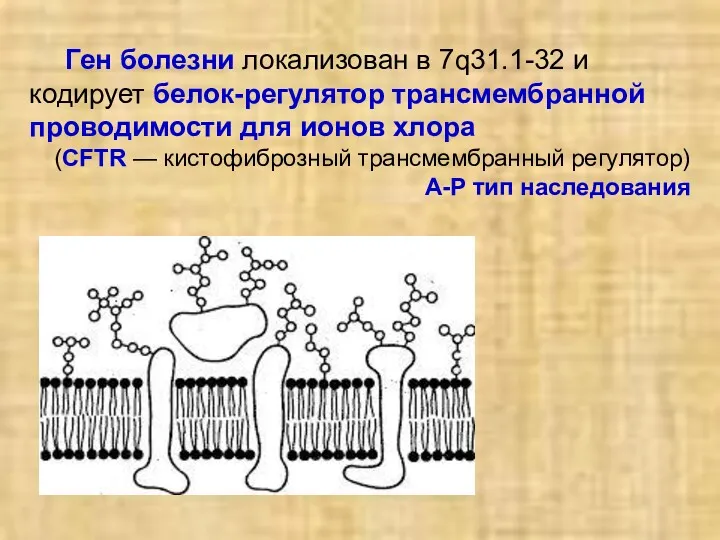
Ген болезни локализован в 7q31.1-32 и кодирует белок-регулятор трансмембранной проводимости для
Ген болезни локализован в 7q31.1-32 и кодирует белок-регулятор трансмембранной проводимости для

Основной патогенетический механизм болезни – увеличение вязкости секрета, выделяемого слизеобразующими железами
Основной патогенетический механизм болезни – увеличение вязкости секрета, выделяемого слизеобразующими железами

Меконеальный илеус
Кишечная непроходимость у новорожденного
Рентгенологическое обследование новорожденного с мекониальной непроходимостью.
Меконеальный илеус
Кишечная непроходимость у новорожденного
Рентгенологическое обследование новорожденного с мекониальной непроходимостью.
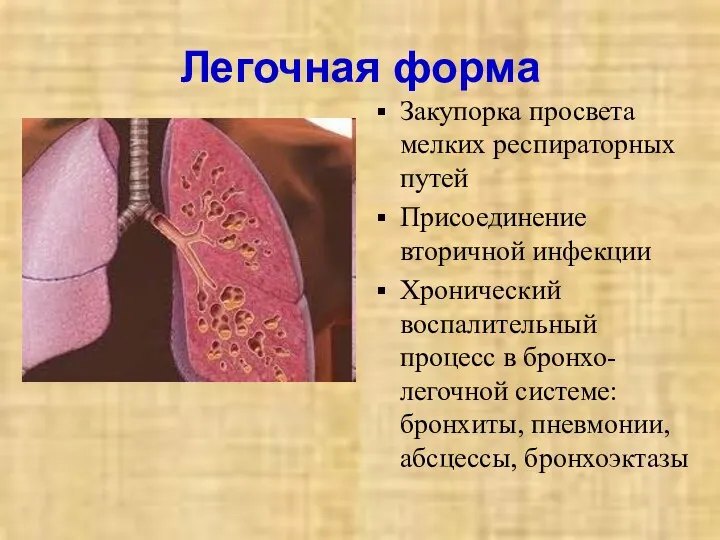
Легочная форма
Закупорка просвета мелких респираторных путей
Присоединение вторичной инфекции
Хронический воспалительный процесс в
Легочная форма
Закупорка просвета мелких респираторных путей
Присоединение вторичной инфекции
Хронический воспалительный процесс в
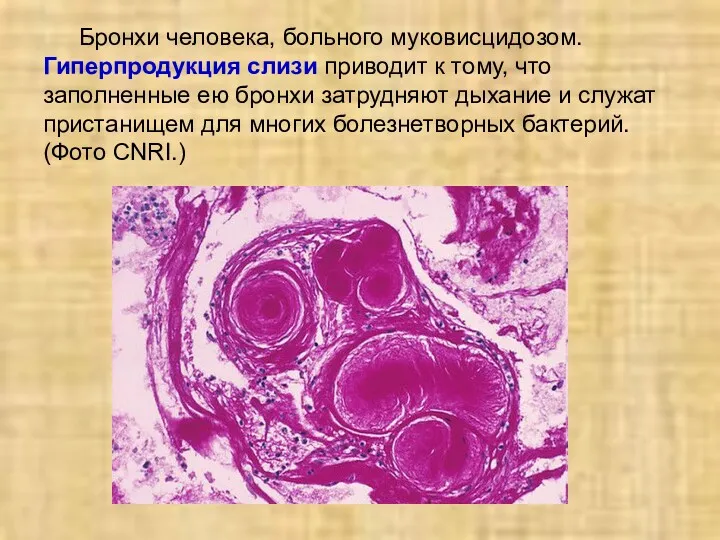
Бронхи человека, больного муковисцидозом. Гиперпродукция слизи приводит к тому, что заполненные
Бронхи человека, больного муковисцидозом. Гиперпродукция слизи приводит к тому, что заполненные

Кишечная форма МВ
Изменение водно-электролитного состава панкреатического сока, его сгущение и затруднение
Кишечная форма МВ
Изменение водно-электролитного состава панкреатического сока, его сгущение и затруднение

Клиническая картина кишечной формы муковисцидоза обусловлена недостаточностью ферментативной активности желудочно-кишечного тракта, которая
Клиническая картина кишечной формы муковисцидоза обусловлена недостаточностью ферментативной активности желудочно-кишечного тракта, которая
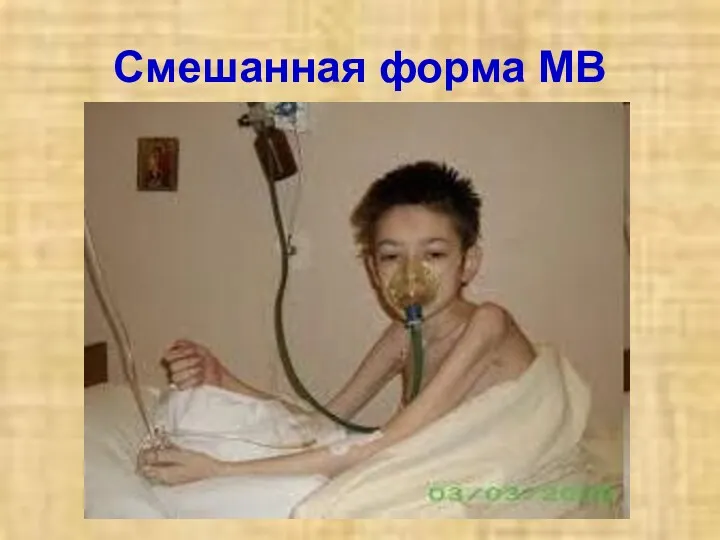
Смешанная форма МВ
Смешанная форма МВ

Неонатальный скрининг –
обследование всех новорожденных с целью раннего (доклинического) выявления
Неонатальный скрининг –
обследование всех новорожденных с целью раннего (доклинического) выявления
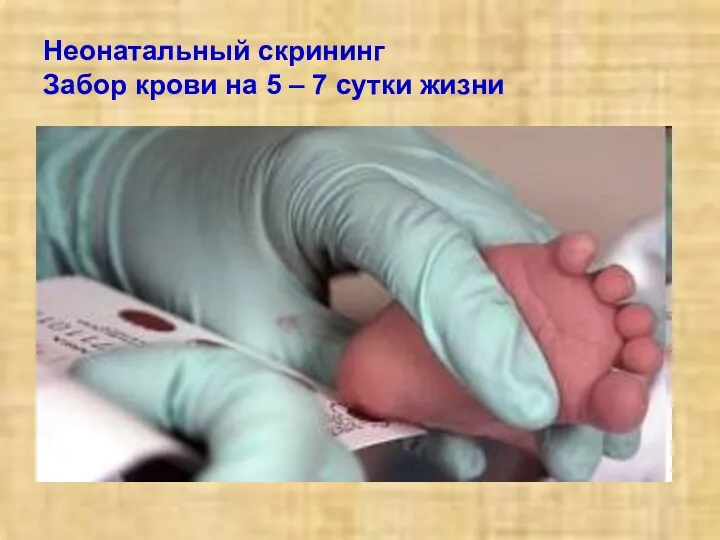
Неонатальный скрининг
Забор крови на 5 – 7 сутки жизни
Неонатальный скрининг
Забор крови на 5 – 7 сутки жизни

ФЕНИЛКЕТОНУРИЯ
ВРОЖДЕННЫЙ ГИПОТИРЕОЗ
ГАЛАКТОЗЕМИЯ
АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ
МУКОВИЗЦИДОЗ
ФЕНИЛКЕТОНУРИЯ
ВРОЖДЕННЫЙ ГИПОТИРЕОЗ
ГАЛАКТОЗЕМИЯ
АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ
МУКОВИЗЦИДОЗ
 Интеграция профилактики передачи ВИЧ от матери к ребенку в эффективный перинатальный уход
Интеграция профилактики передачи ВИЧ от матери к ребенку в эффективный перинатальный уход Жүректің өткізгіштік жүйесінің морфофункциональдық ерекшеліктері
Жүректің өткізгіштік жүйесінің морфофункциональдық ерекшеліктері Патология теплового обмена
Патология теплового обмена Состояние зрительных функций в возрастном аспекте
Состояние зрительных функций в возрастном аспекте Лабораторные методы исследования
Лабораторные методы исследования Хроматография әдісінің негізі
Хроматография әдісінің негізі Антиаритмические препараты их классификация и механизм действия
Антиаритмические препараты их классификация и механизм действия Ларинготрахеит у детей
Ларинготрахеит у детей Жидкие лекарственные формы
Жидкие лекарственные формы Отравления алкоголем и его суррогатами. Первая помощь, интенсивная терапия
Отравления алкоголем и его суррогатами. Первая помощь, интенсивная терапия Жаңа туған нәрестенің физиологиясы және патологиясы
Жаңа туған нәрестенің физиологиясы және патологиясы Гипофиз. Надпочечники. Половые железы. Лекция № 32
Гипофиз. Надпочечники. Половые железы. Лекция № 32 Приготовление питательной эмульсии для рук и ногтей
Приготовление питательной эмульсии для рук и ногтей Инфекции, передаваемые половым путем. Меры их профилактики
Инфекции, передаваемые половым путем. Меры их профилактики ЛП, влияющие на функции органов дыхания
ЛП, влияющие на функции органов дыхания Состояние условий труда на предприятиях Пермского края. Сохранение здоровья работающих
Состояние условий труда на предприятиях Пермского края. Сохранение здоровья работающих Нетуберкулёзные микобактерии (НТМ). Диагностика и лечение
Нетуберкулёзные микобактерии (НТМ). Диагностика и лечение Основная медицинская помощь новорожденному
Основная медицинская помощь новорожденному Медицина и общество
Медицина и общество Теплолечение. Санаторно-курортное лечение. Лекция № 4
Теплолечение. Санаторно-курортное лечение. Лекция № 4 Эмбриология ғылымының дамуы
Эмбриология ғылымының дамуы Дизартрия. Формы дизартрии
Дизартрия. Формы дизартрии Основные врожденные заболевания ОДС
Основные врожденные заболевания ОДС Гемопоэз и иммунный ответ
Гемопоэз и иммунный ответ Жүрек-қан тамырлар жүйесінің аурулары. Жүрек ақауы
Жүрек-қан тамырлар жүйесінің аурулары. Жүрек ақауы История развития Всероссийской службы медицины катастроф (ВСМК). Определение, задачи и основные принципы организации помощи
История развития Всероссийской службы медицины катастроф (ВСМК). Определение, задачи и основные принципы организации помощи Ісік, тін өсіндісі, бластома (tumoz)
Ісік, тін өсіндісі, бластома (tumoz) История воспаления - это история медицины
История воспаления - это история медицины