- Мукополисахаридоз

Содержание
- 2. Мукополисахаридоз I типа синдром Гурлер. Мукополисахаридоз типа IH или синдром Гурлер встречается у 1 новорожденного из
- 3. Рис. 1. Синдром Гурлер: типичные внешние проявления. Для данной формы мукополисахаридоза характерны грубые черты лица и
- 5. Рис. 2б). Рентгенологические признаки синдрома Гурлер — деформация таза и бедренных костей.
- 7. Рис. 2в). Рентгенологические признаки синдрома Гурлер — характерный вид кистей.
- 8. Мукополисахаридоз II тип Гунтера. Мукополисахаридоз II типа До 3-6 лет развитие детей соответствует норме. Первым признаком
- 9. Рис. 3б). Синдром Гунтера у мальчика 2 лет — изменения скелета слабо выражены, нет кифоза, контрактур.
- 10. Помутнение роговицы . Мукополисахаридозы
- 11. Мукополисахаридоз III типа -синдром Санфилиппо. Описан американским педиатром Санфилиппо (S.J. Sanfilippo) в 1963 г. Частота 1
- 12. Мукополисахаридоз типа IV- снидром Моркио. описана в 1929 г. уругвайским педиатром Моркио наблюдается у 1 новорожденного
- 13. Рис. 5. Синдром Моркио: типичные внешние проявления.
- 14. Рис. 6в). Рентгенологические признаки синдрома Моркио — деформация костей кисти.
- 15. V-тип.синдрома Шейне Для синдрома Шейе характерен низкий рост, уплощенная переносица, короткая шея, контрактуры суставов, гипотония мышц
- 16. Рис. 4. Болезнь Шейе: типичные внешние проявления
- 17. Тип VI, VII, VIII. Мукополисахаридоз типа VI или болезнь Марото-Лами (описана в 1960 г. французами Лами
- 18. Рис. 7а). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — грубые черты
- 19. Рис. 7б). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — контрактуры верхних
- 20. Рис. 8а). Рентгенологические признаки синдрома Марото — Лами — изменения позвоночника.
- 21. Рис. 8б). Рентгенологические признаки синдрома Марото — Лами — деформация костей таза.
- 22. Диагностика Диагностика мукополисахаридоза основывается на его характерных проявлениях, результатах рентгенологического исследования, установления экскреции гликозаминогликанов с мочой,
- 23. Причины. Причиной развития мукополисахаридоза являетсня нарушение ферментативного катализа гликозаминогликанов в лизосомах. нарушается процесс расщепления и сохранения
- 24. Лечение. Лечение симптоматическое. При этом больных наблюдают разные специалисты — хирурги (удаление грыж), ортопеды (ортопедическая коррекция
- 26. Скачать презентацию

Мукополисахаридоз I типа синдром Гурлер.
Мукополисахаридоз типа IH или синдром Гурлер встречается
Мукополисахаридоз I типа синдром Гурлер.
Мукополисахаридоз типа IH или синдром Гурлер встречается
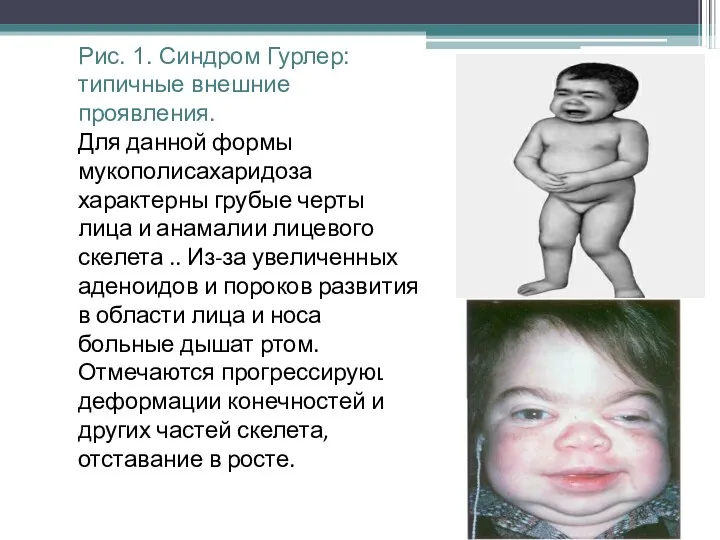
Рис. 1. Синдром Гурлер: типичные внешние проявления.
Для данной формы мукополисахаридоза
Рис. 1. Синдром Гурлер: типичные внешние проявления.
Для данной формы мукополисахаридоза

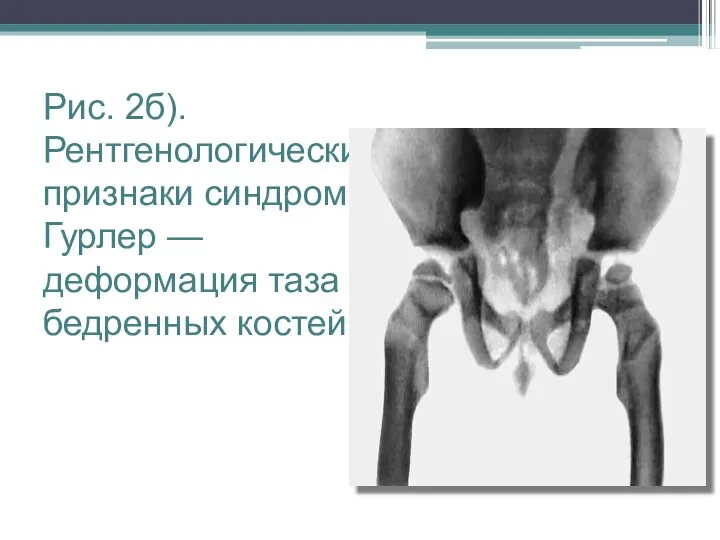
Рис. 2б). Рентгенологические признаки синдрома Гурлер — деформация таза и бедренных костей.
Рис. 2б). Рентгенологические признаки синдрома Гурлер — деформация таза и бедренных костей.
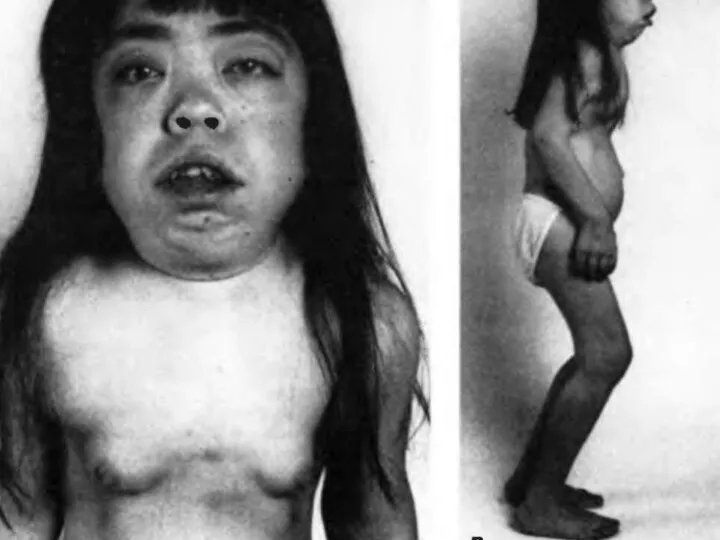
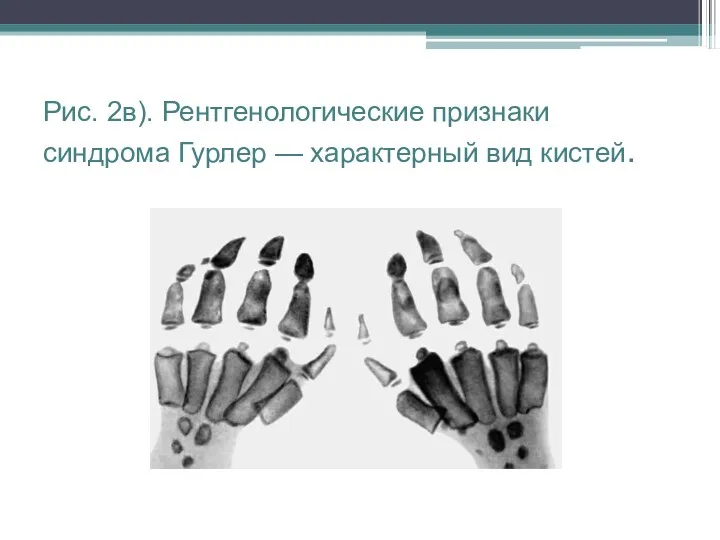
Рис. 2в). Рентгенологические признаки синдрома Гурлер — характерный вид кистей.
Рис. 2в). Рентгенологические признаки синдрома Гурлер — характерный вид кистей.

Мукополисахаридоз II тип Гунтера.
Мукополисахаридоз II типа До 3-6 лет развитие
Мукополисахаридоз II тип Гунтера.
Мукополисахаридоз II типа До 3-6 лет развитие

Рис. 3б). Синдром Гунтера у мальчика 2 лет — изменения скелета слабо
Рис. 3б). Синдром Гунтера у мальчика 2 лет — изменения скелета слабо
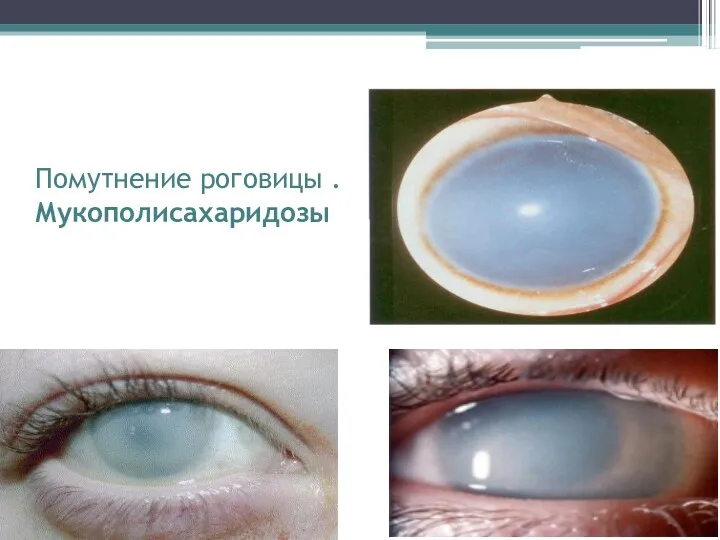
Помутнение роговицы .
Мукополисахаридозы
Помутнение роговицы .
Мукополисахаридозы

Мукополисахаридоз III типа -синдром Санфилиппо.
Описан американским педиатром Санфилиппо (S.J. Sanfilippo) в 1963 г.
Мукополисахаридоз III типа -синдром Санфилиппо.
Описан американским педиатром Санфилиппо (S.J. Sanfilippo) в 1963 г.

Мукополисахаридоз типа IV- снидром Моркио.
описана в 1929 г. уругвайским педиатром
Мукополисахаридоз типа IV- снидром Моркио.
описана в 1929 г. уругвайским педиатром

Рис. 5. Синдром Моркио: типичные внешние проявления.
Рис. 5. Синдром Моркио: типичные внешние проявления.
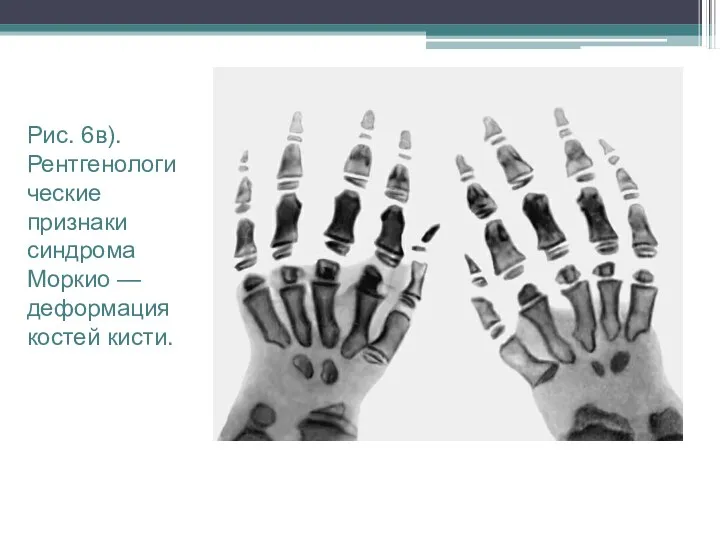
Рис. 6в). Рентгенологические признаки синдрома Моркио — деформация костей кисти.
Рис. 6в). Рентгенологические признаки синдрома Моркио — деформация костей кисти.

V-тип.синдрома Шейне
Для синдрома Шейе характерен низкий рост, уплощенная переносица, короткая
V-тип.синдрома Шейне
Для синдрома Шейе характерен низкий рост, уплощенная переносица, короткая
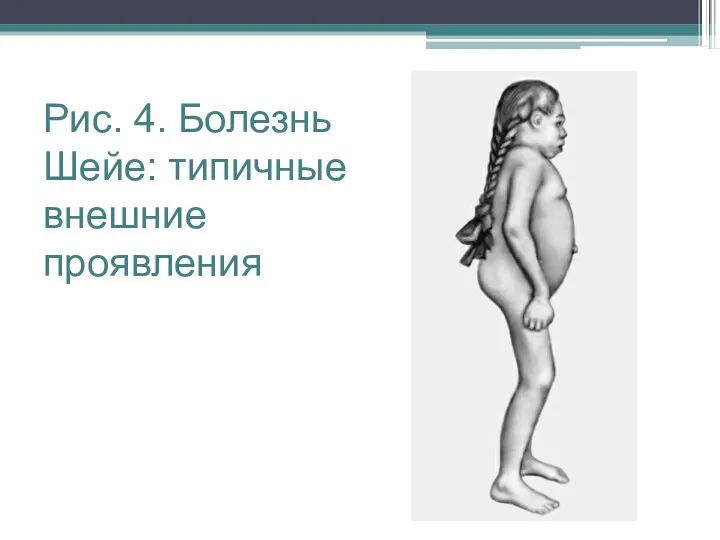
Рис. 4. Болезнь Шейе: типичные внешние проявления
Рис. 4. Болезнь Шейе: типичные внешние проявления

Тип VI, VII, VIII.
Мукополисахаридоз типа VI или болезнь Марото-Лами (описана в
Тип VI, VII, VIII.
Мукополисахаридоз типа VI или болезнь Марото-Лами (описана в
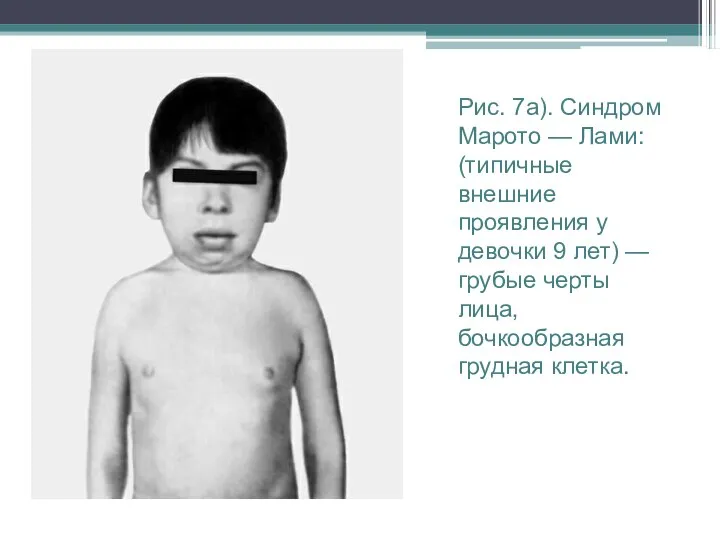
Рис. 7а). Синдром Марото — Лами: (типичные внешние проявления у девочки 9
Рис. 7а). Синдром Марото — Лами: (типичные внешние проявления у девочки 9

Рис. 7б). Синдром Марото — Лами: (типичные внешние проявления у девочки 9
Рис. 7б). Синдром Марото — Лами: (типичные внешние проявления у девочки 9
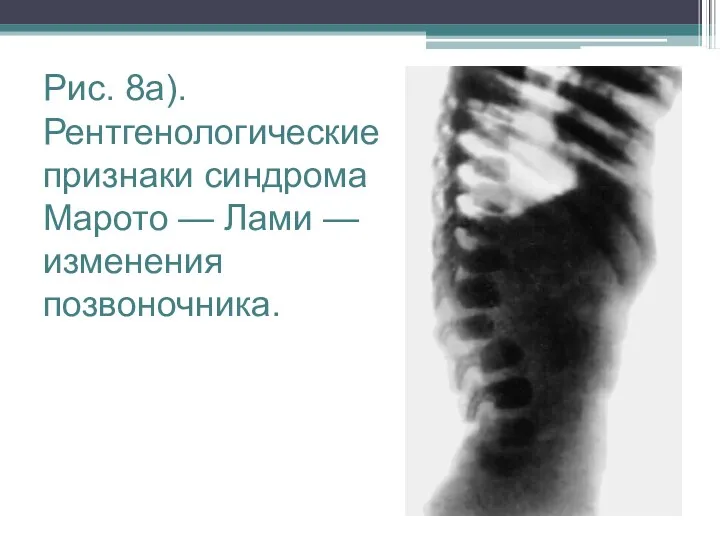
Рис. 8а). Рентгенологические признаки синдрома Марото — Лами — изменения позвоночника.
Рис. 8а). Рентгенологические признаки синдрома Марото — Лами — изменения позвоночника.
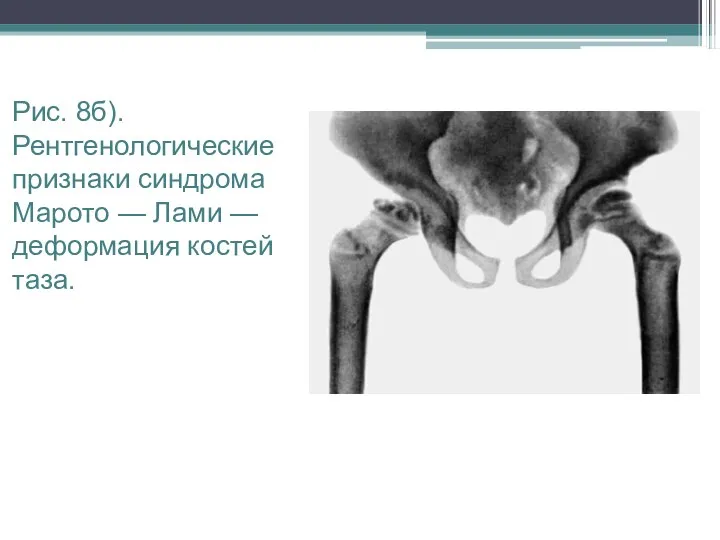
Рис. 8б). Рентгенологические признаки синдрома Марото — Лами — деформация костей таза.
Рис. 8б). Рентгенологические признаки синдрома Марото — Лами — деформация костей таза.

Диагностика
Диагностика мукополисахаридоза основывается на его характерных проявлениях, результатах рентгенологического исследования, установления
Диагностика
Диагностика мукополисахаридоза основывается на его характерных проявлениях, результатах рентгенологического исследования, установления

Причины.
Причиной развития мукополисахаридоза являетсня нарушение ферментативного катализа гликозаминогликанов в лизосомах. нарушается
Причины.
Причиной развития мукополисахаридоза являетсня нарушение ферментативного катализа гликозаминогликанов в лизосомах. нарушается

Лечение.
Лечение симптоматическое. При этом больных наблюдают разные специалисты — хирурги (удаление грыж),
Лечение.
Лечение симптоматическое. При этом больных наблюдают разные специалисты — хирурги (удаление грыж),
 Головные вши (Pediculus humanus capitis)
Головные вши (Pediculus humanus capitis) Гипоплазия эмали: этиология, патогенез, клиника, диагностика, лечение. Наследственные пороки развития твердых тканей зубов
Гипоплазия эмали: этиология, патогенез, клиника, диагностика, лечение. Наследственные пороки развития твердых тканей зубов Көз туберкулезі
Көз туберкулезі Анализ результатов лечения гидроцефалии у детей
Анализ результатов лечения гидроцефалии у детей Основные принципы асептики в терапевтической стоматологии. Инфекционный контроль
Основные принципы асептики в терапевтической стоматологии. Инфекционный контроль Организация ортопедического отделения. Зуботехническая лаборатория
Организация ортопедического отделения. Зуботехническая лаборатория Лечебная физкультура
Лечебная физкультура Стационарлық көмек деңгейіндегі заманауи автоматтандырылған деректер базасымен таныстыру
Стационарлық көмек деңгейіндегі заманауи автоматтандырылған деректер базасымен таныстыру Обеспечение безопасного пространства для пациента и персонала в медицинских организациях
Обеспечение безопасного пространства для пациента и персонала в медицинских организациях Рациональное питание
Рациональное питание Закаливание организма
Закаливание организма Служение больницы и учреждения. Анонимные Наркоманы
Служение больницы и учреждения. Анонимные Наркоманы Одонтогенді гайморит
Одонтогенді гайморит Firearm injuries
Firearm injuries Прививка против гриппа
Прививка против гриппа Особенности сестринского ухода в гериатрии. Болезни органов дыхания у гериатрических пациентов
Особенности сестринского ухода в гериатрии. Болезни органов дыхания у гериатрических пациентов Компенсаторно-приспособительные реакции
Компенсаторно-приспособительные реакции Правила постановки периферического венозного катетера
Правила постановки периферического венозного катетера Тағамдық және санитарлы микробиология
Тағамдық және санитарлы микробиология Особо опасные инфекции (ООИ)
Особо опасные инфекции (ООИ) Гангрена, некроз, язвы, свищи
Гангрена, некроз, язвы, свищи Жүктілік кезіндегі гипертензивті жағдайлар
Жүктілік кезіндегі гипертензивті жағдайлар Дүниежүзілік денсаулық сақтау ұйымы (ДДҰ) Қазақстанда
Дүниежүзілік денсаулық сақтау ұйымы (ДДҰ) Қазақстанда Оқу кестелері мен муляждарды қолдану арқылы ауыз қуысы мен ауыз қуысы құрылымдарының функцияларын зерттеу
Оқу кестелері мен муляждарды қолдану арқылы ауыз қуысы мен ауыз қуысы құрылымдарының функцияларын зерттеу Патогенна дія хімічних та біологічних факторів на організм. Роль спадковості в патології. (Лекція 4)
Патогенна дія хімічних та біологічних факторів на організм. Роль спадковості в патології. (Лекція 4) Средства, влияющие на афферентную нервную систему
Средства, влияющие на афферентную нервную систему Хірургічні захворювання прямої кишки
Хірургічні захворювання прямої кишки Алгоритм манипуляций по оказанию больным первой медицинской помощи при возникновении неотложных ситуаций
Алгоритм манипуляций по оказанию больным первой медицинской помощи при возникновении неотложных ситуаций