- Наследственно-дегенеративные заболевания нервной системы

Содержание
- 2. Наследственные заболевания – это большая группа болезней, обусловленных изменениями наследственной информации Наука, дисциплина, изучающая наследственные болезни,
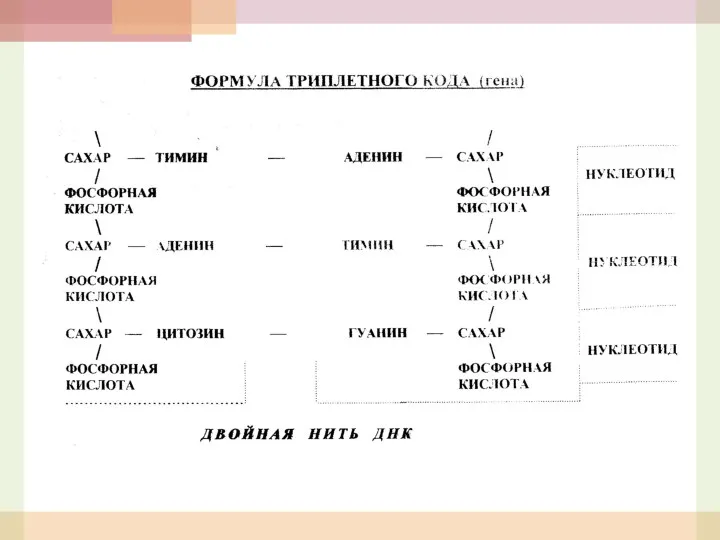
- 3. Формула наследственных болезней Один мутантный ген – один белок или фермент – одна болезнь. Материальными носителями
- 4. Человеческий кариотип (мужчина)
- 5. Хромосомы состоят из двойной нити ДНК – цепочка из сочетаний молекулы сахара (дезоксирибозы), фосфорной кислоты и
- 6. Участок хромосомы, т.е. нити ДНК, состоящий из трех нуклеотидов и определяющий одну функцию – синтез определенного
- 8. В настоящее время геном человека раскрыт. Во всей нити ДНК, расположенного в 46 хромосомах человека, насчитывается
- 9. Патогенез наследственных болезней Для того, чтобы возникла мутация, достаточно замены одной пары нуклеотида на другую в
- 10. В результате подобной ломки возникает дефицит того или иного биологически активного вещества, что сопровождается извращением образования
- 11. Наследственная патология может определяться не только мутацией в одном гене, но и комбинацией двух или нескольких
- 12. Среди наследственных заболеваний с вовлечением структур нервной системы выделяют в основном следующие группы: Наследственные системные дегенерации
- 13. I. Наследственные системные дегенерации нервной системы Для этой группы болезней нервной системы характерно прогрессирующее течение с
- 14. При многих системных дегенерациях отмечается сочетанность поражения структур нервной системы и внутренних органов, кожных покровов, а
- 15. Семейный спастический паралич Штрюмпеля Ядром клинической картины является прогрессирующий нижний спастический парапарез. До 70% заболеваний протекает
- 16. Изолированные формы наследственной спастической параплегии с аутосомно-доминантным наследованием обусловлены повреждением большого количества генов в различных хромосомных
- 17. Все эти белки имеют прямое отношение к механизмам аксонального транспорта пирамидных клеток Беца. В результате повреждения
- 18. С учетом отмеченного, при спастической параплегии отмечается дегенерация пирамидных трактов боковых столбов спинного мозга, более выраженная
- 19. Клиника. Первые симптомы могут проявиться в любом возрасте, чаще до 10-15 лет: скованность и быстрая утомляемость
- 20. Лечение. Назначение препаратов, направленных на снижение спастичности: баклофен 10-30 мг/сут, сирдалуд до 20 мг/сут, а также
- 21. Атаксия Фридрейха Заболевание наследуется по аутосомно-рецессив-ному типу. Частота: 2-10 больных на 100 000 населения. Мутантный ген
- 22. Относится к спинальным формам наследственных атаксий. Дегенеративным процессом захватываются задние и боковые столбы спинного мозга, приводя
- 23. В качестве одного из первых симптомов больные отмечают неуверенность при ходьбе (особенно в темноте), пошатывание и
- 24. Лечение: симптоматическая терапия, физические методы, ортопедические мероприятия, заместительная терапия при дефиците витамина Е, В1 и В6
- 25. II. Наследственные болезни обмена, протекающие с поражением нервной системы В эту группу болезней можно отнести: гепато-лентикулярная
- 26. Гепато-лентикулярная дегенерация (болезнь Вильсона – Коновалова) В организме возникает дефицит белкового вещества – церулоплазмина, участвующего в
- 27. В головном мозге медь откладывается преимущественно в экстрапирамидных базальных ядрах, приводя к дегенерации мозговых структур и
- 28. Клиническая картина. Заболевание начинает проявляться в 10 - 15 лет, характеризуется нарастающей мышечной ригидностью, разнообразными гиперкинезами
- 29. Медь откладывается и в роговице, образуя кольцо Кайзера-Флейшера (золотисто-зеленого или зеленовато-коричневого цвета) и коже (зуд). Нарушена
- 30. Лечение. Препараты, способствующие выделению из организма меди (унитиол, пеницилламин, купренил). Препараты, снижающие мышечный тонус (циклодол, мидокалм).
- 31. Фенилкетонурия (фенилпировиноградная олигофрения) Пища в желудочно-кишечном тракте распадается до аминокислот и воды. Всасываясь в кровеносную систему
- 32. При фенилкетонурии, образующаяся из пищи аминокислота фенилаланин расщепляется частично, т.к. фермент фенилаланиноксидаза вырабатывается в организме в
- 33. Клиника. Заболевание начинает проявляться у здорового ребенка в 5–7-месячном возрасте: рвота, судороги, двигательные беспокойства, пирамидные знаки,
- 34. Больной фенилкетонурией в «позах портного».
- 35. Дети часто белокурые со светлой кожей и голубыми глазами. Диагностирующим тестом является проба Феллинга: в моче
- 36. III. Факоматозы. Это группа заболеваний, при которых отмечается поражение кожных покровов с образованием пигментированных и непигменти-рованных
- 37. Характерными симптомами заболеваний являются пигментированные или депигментированные пятна на коже, ангиомы сосудов, фибромы и ряд других
- 38. Энцефалотригеминальный ангиоматоз Штурге-Вебера Передается аутосомно-доминантно с весьма низкой пенетрантностью. Типичными являются ангиома кожных покровов, эпилептиформные припадки
- 39. Нейрофиброматоз Реклингаузена Первое проявление болезни наблюдается в подростковом возрасте: на фоне пигментных пятен кожи (кофе с
- 40. IV. Наследственные нервно-мышечные заболевания Это большая группа патологических состояний. Ядром клинической симптоматики является неуклонно прогрессирующая или
- 41. Принято выделять 6 больших подгрупп нервно-мышечных заболеваний: Первично-прогрессирующие мышечные дистрофии – миопатии. Первично-непрогрессирующие доброкачественные миопатии. Вторично-прогрессирующие
- 42. При всех формах наследственных нервно-мышечных заболеваний трагедия разыгрывается в пределах так называемой двигательной единицы, введенной в
- 43. В ниже приведенной таблице представлены наиболее часто встречающиеся формы нервно-мышечных заболеваний. Миопатии А. Прогрессирующие формы: а)
- 44. II. Неврогенные амиотрофии. А. Спинальные формы: а) детская амиотрофия Верднига – Гофмана; б) юношеская амиотрофия Кугельберга
- 45. Патогенез первично-прогрессирующих мышечных дистрофий – миопатий Можно считать уже доказанным, что у больных миопатией синтез белка
- 46. У больных миопатией отмечается усиленное выделение с мочой таких аминокислот как креатин и креатинин. В норме
- 47. Наряду с измененными мышечными волокнами вперемешку находятся нормальные. Такая мышца внешне гипертрофирована, но бессильная. Это и
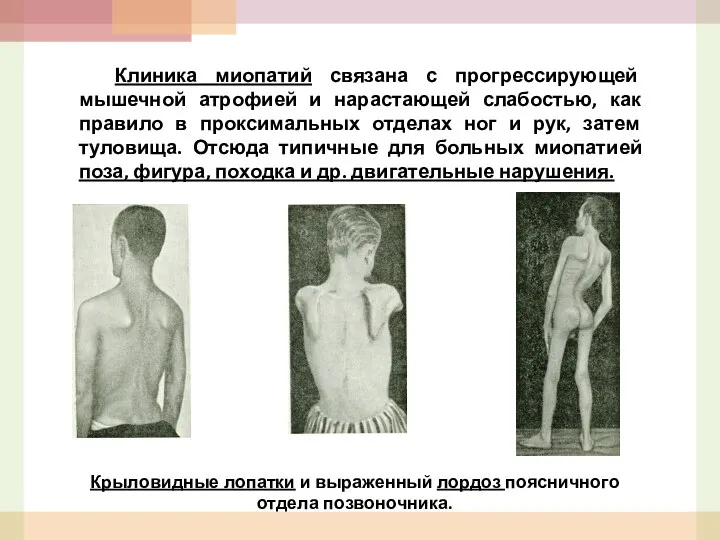
- 48. Клиника миопатий связана с прогрессирующей мышечной атрофией и нарастающей слабостью, как правило в проксимальных отделах ног
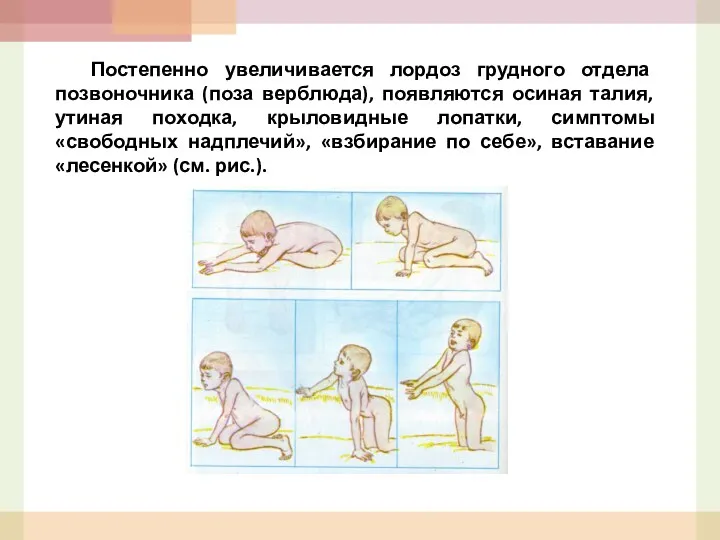
- 49. Постепенно увеличивается лордоз грудного отдела позвоночника (поза верблюда), появляются осиная талия, утиная походка, крыловидные лопатки, симптомы
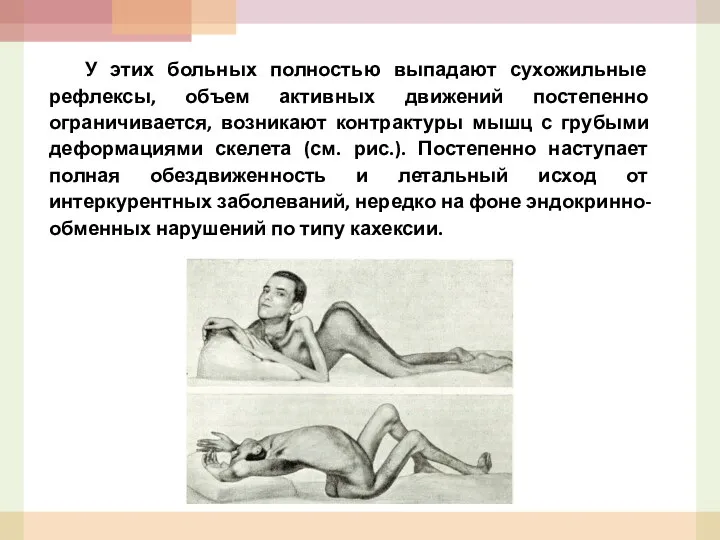
- 50. У этих больных полностью выпадают сухожильные рефлексы, объем активных движений постепенно ограничивается, возникают контрактуры мышц с
- 51. Рассмотрим кратко ряд клинических форм заболеваний: а) Псевдогипертрофическая амиотрофия Дюшена. Болезнь начинает проявляться к 3-4 годам,
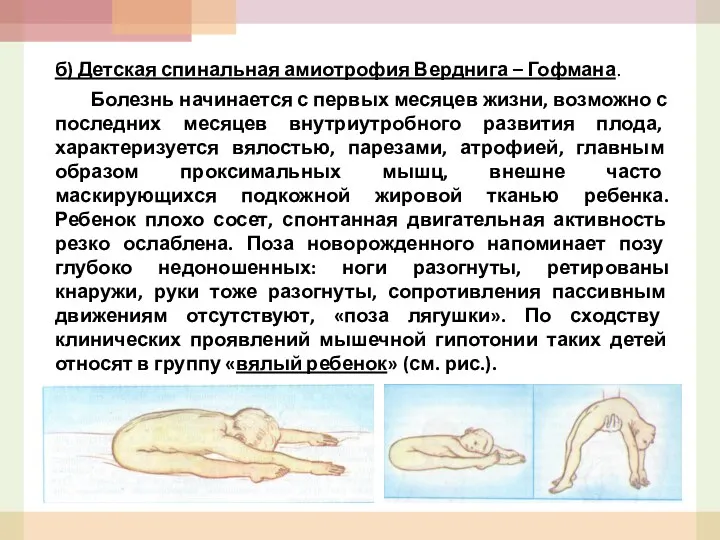
- 52. б) Детская спинальная амиотрофия Верднига – Гофмана. Болезнь начинается с первых месяцев жизни, возможно с последних
- 53. В пораженных мышцах наблюдаются фибриллярные и фасцикулярные подергивания. Уточнению диагноза способствует электромиографическое исследование, при котором выявляются
- 54. в) Невральная амиотрофия Шарко – Мари. Заболевание насле-дуется чаще по аутосомно-доминантному типу, начи-нается в детском и
- 55. Наряду с двигательными нарушениями для этих больных характерны чувствительные расстройства по типу «носков», «чулков» и «перчаток».
- 56. г) Миастения. Основным симптомом заболевания является нарастающая мышечная слабость, резко усиливающаяся при повторных активных движениях: нарастающий
- 57. Специальные исследования показали нарушение синтеза ацетилхолина при избытке холинэстеразы в тканях больных миастенией. У больных, погибших
- 58. При миастенических кризах (резкое нарастание симптомов заболевания) рекомендуется срочное введение 0,5 мл 0,05% раствора прозерина в/в
- 60. Скачать презентацию

Наследственные заболевания – это большая группа болезней, обусловленных изменениями наследственной
Наследственные заболевания – это большая группа болезней, обусловленных изменениями наследственной

Формула наследственных болезней
Один мутантный ген – один белок или фермент –
Формула наследственных болезней
Один мутантный ген – один белок или фермент –
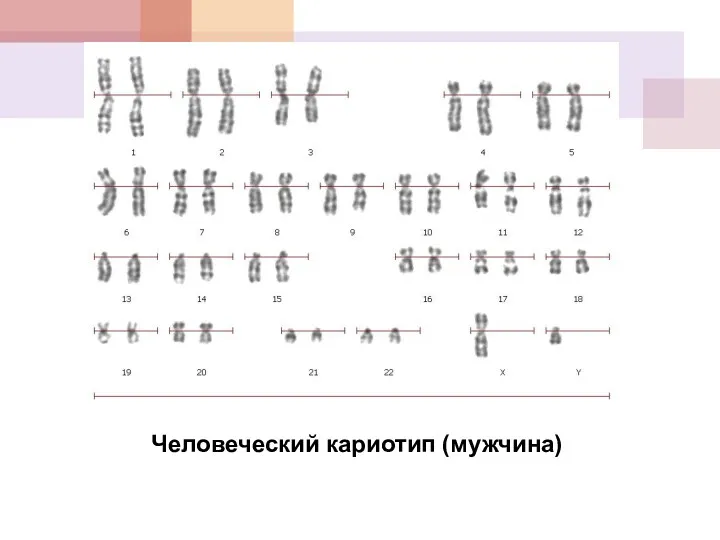
Человеческий кариотип (мужчина)
Человеческий кариотип (мужчина)
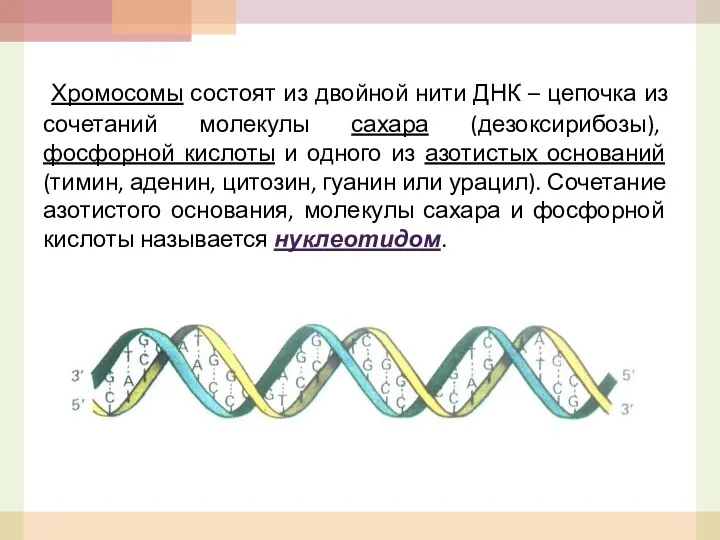
Хромосомы состоят из двойной нити ДНК – цепочка из сочетаний
Хромосомы состоят из двойной нити ДНК – цепочка из сочетаний

Участок хромосомы, т.е. нити ДНК, состоящий из трех нуклеотидов и
Участок хромосомы, т.е. нити ДНК, состоящий из трех нуклеотидов и

В настоящее время геном человека раскрыт. Во всей нити ДНК,
В настоящее время геном человека раскрыт. Во всей нити ДНК,

Патогенез наследственных болезней
Для того, чтобы возникла мутация, достаточно замены
Патогенез наследственных болезней
Для того, чтобы возникла мутация, достаточно замены

В результате подобной ломки возникает дефицит того или иного биологически активного
В результате подобной ломки возникает дефицит того или иного биологически активного

Наследственная патология может определяться не только мутацией в одном гене, но
Наследственная патология может определяться не только мутацией в одном гене, но

Среди наследственных заболеваний с вовлечением структур нервной системы выделяют в основном
Среди наследственных заболеваний с вовлечением структур нервной системы выделяют в основном

I. Наследственные системные дегенерации нервной системы
Для этой группы болезней нервной системы
I. Наследственные системные дегенерации нервной системы
Для этой группы болезней нервной системы

При многих системных дегенерациях отмечается сочетанность поражения структур нервной системы и
При многих системных дегенерациях отмечается сочетанность поражения структур нервной системы и

Семейный спастический паралич Штрюмпеля
Ядром клинической картины является прогрессирующий нижний спастический парапарез.
До
Семейный спастический паралич Штрюмпеля
Ядром клинической картины является прогрессирующий нижний спастический парапарез.
До

Изолированные формы наследственной спастической параплегии с аутосомно-доминантным наследованием обусловлены повреждением большого
Изолированные формы наследственной спастической параплегии с аутосомно-доминантным наследованием обусловлены повреждением большого

Все эти белки имеют прямое отношение к механизмам аксонального транспорта пирамидных
Все эти белки имеют прямое отношение к механизмам аксонального транспорта пирамидных

С учетом отмеченного, при спастической параплегии отмечается дегенерация пирамидных трактов боковых
С учетом отмеченного, при спастической параплегии отмечается дегенерация пирамидных трактов боковых

Клиника. Первые симптомы могут проявиться в любом возрасте, чаще до 10-15
Клиника. Первые симптомы могут проявиться в любом возрасте, чаще до 10-15

Лечение. Назначение препаратов, направленных на снижение спастичности: баклофен 10-30 мг/сут, сирдалуд
Лечение. Назначение препаратов, направленных на снижение спастичности: баклофен 10-30 мг/сут, сирдалуд

Атаксия Фридрейха
Заболевание наследуется по аутосомно-рецессив-ному типу. Частота: 2-10 больных на 100
Атаксия Фридрейха
Заболевание наследуется по аутосомно-рецессив-ному типу. Частота: 2-10 больных на 100

Относится к спинальным формам наследственных атаксий.
Дегенеративным процессом захватываются задние и боковые
Относится к спинальным формам наследственных атаксий.
Дегенеративным процессом захватываются задние и боковые

В качестве одного из первых симптомов больные отмечают неуверенность при ходьбе
В качестве одного из первых симптомов больные отмечают неуверенность при ходьбе

Лечение:
симптоматическая терапия,
физические методы,
ортопедические мероприятия,
заместительная терапия при дефиците
Лечение:
симптоматическая терапия,
физические методы,
ортопедические мероприятия,
заместительная терапия при дефиците

II. Наследственные болезни обмена, протекающие с поражением нервной системы
В эту
II. Наследственные болезни обмена, протекающие с поражением нервной системы
В эту

Гепато-лентикулярная дегенерация (болезнь Вильсона – Коновалова)
В организме возникает дефицит белкового вещества
Гепато-лентикулярная дегенерация (болезнь Вильсона – Коновалова)
В организме возникает дефицит белкового вещества

В головном мозге медь откладывается преимущественно в экстрапирамидных базальных ядрах,
В головном мозге медь откладывается преимущественно в экстрапирамидных базальных ядрах,

Клиническая картина. Заболевание начинает проявляться в 10 - 15 лет, характеризуется
Клиническая картина. Заболевание начинает проявляться в 10 - 15 лет, характеризуется
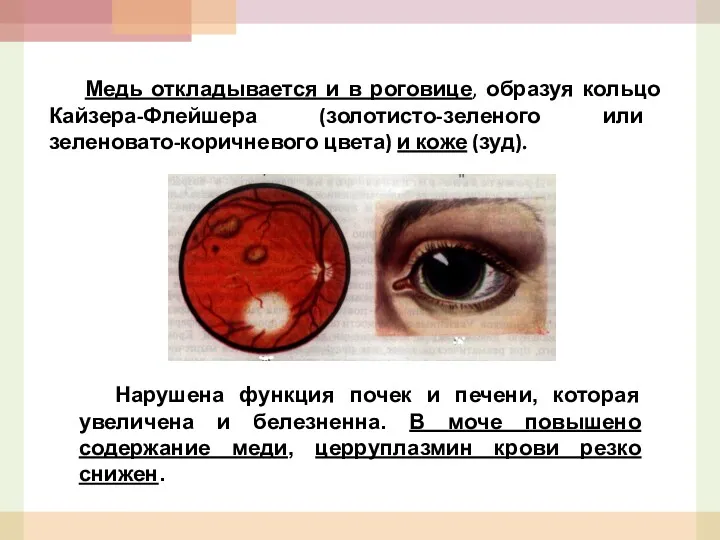
Медь откладывается и в роговице, образуя кольцо Кайзера-Флейшера (золотисто-зеленого или зеленовато-коричневого
Медь откладывается и в роговице, образуя кольцо Кайзера-Флейшера (золотисто-зеленого или зеленовато-коричневого

Лечение.
Препараты, способствующие выделению из организма меди (унитиол, пеницилламин, купренил).
Препараты, снижающие
Лечение.
Препараты, способствующие выделению из организма меди (унитиол, пеницилламин, купренил).
Препараты, снижающие

Фенилкетонурия
(фенилпировиноградная олигофрения)
Пища в желудочно-кишечном тракте распадается до аминокислот и воды. Всасываясь
Фенилкетонурия
(фенилпировиноградная олигофрения)
Пища в желудочно-кишечном тракте распадается до аминокислот и воды. Всасываясь

При фенилкетонурии, образующаяся из пищи аминокислота фенилаланин расщепляется частично, т.к. фермент
При фенилкетонурии, образующаяся из пищи аминокислота фенилаланин расщепляется частично, т.к. фермент

Клиника. Заболевание начинает проявляться у здорового ребенка в 5–7-месячном возрасте: рвота,
Клиника. Заболевание начинает проявляться у здорового ребенка в 5–7-месячном возрасте: рвота,
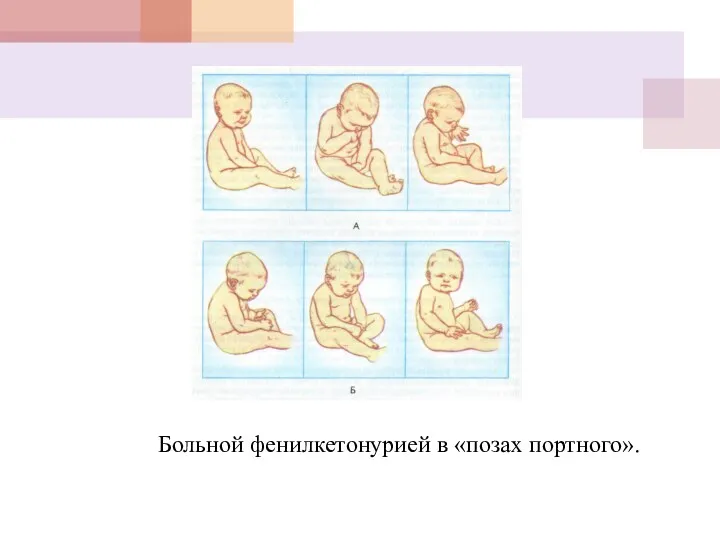
Больной фенилкетонурией в «позах портного».
Больной фенилкетонурией в «позах портного».

Дети часто белокурые со светлой кожей и голубыми глазами. Диагностирующим тестом
Дети часто белокурые со светлой кожей и голубыми глазами. Диагностирующим тестом

III. Факоматозы.
Это группа заболеваний, при которых отмечается поражение кожных покровов с
III. Факоматозы.
Это группа заболеваний, при которых отмечается поражение кожных покровов с

Характерными симптомами заболеваний являются пигментированные или депигментированные пятна на коже, ангиомы
Характерными симптомами заболеваний являются пигментированные или депигментированные пятна на коже, ангиомы
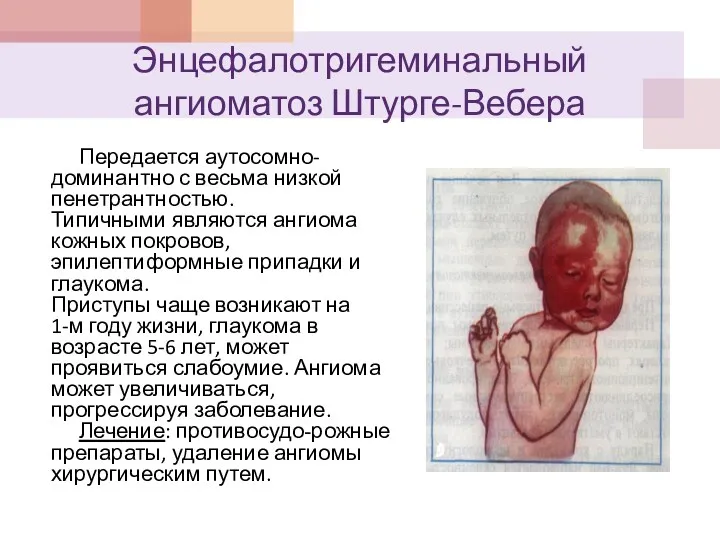
Энцефалотригеминальный ангиоматоз Штурге-Вебера
Передается аутосомно-доминантно с весьма низкой пенетрантностью.
Типичными являются ангиома кожных
Энцефалотригеминальный ангиоматоз Штурге-Вебера
Передается аутосомно-доминантно с весьма низкой пенетрантностью.
Типичными являются ангиома кожных

Нейрофиброматоз Реклингаузена
Первое проявление болезни наблюдается в подростковом возрасте: на фоне пигментных
Нейрофиброматоз Реклингаузена
Первое проявление болезни наблюдается в подростковом возрасте: на фоне пигментных

IV. Наследственные нервно-мышечные заболевания
Это большая группа патологических состояний. Ядром клинической симптоматики
IV. Наследственные нервно-мышечные заболевания
Это большая группа патологических состояний. Ядром клинической симптоматики

Принято выделять 6 больших подгрупп нервно-мышечных заболеваний:
Первично-прогрессирующие мышечные дистрофии –
Принято выделять 6 больших подгрупп нервно-мышечных заболеваний:
Первично-прогрессирующие мышечные дистрофии –
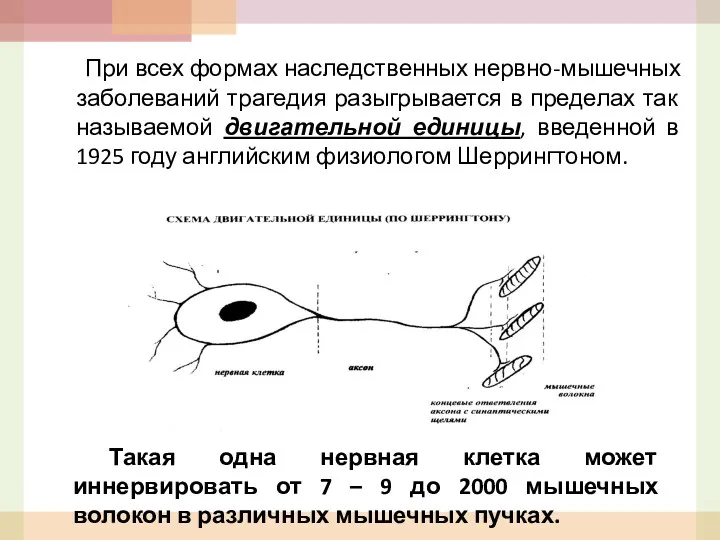
При всех формах наследственных нервно-мышечных заболеваний трагедия разыгрывается в пределах
При всех формах наследственных нервно-мышечных заболеваний трагедия разыгрывается в пределах

В ниже приведенной таблице представлены наиболее часто встречающиеся формы нервно-мышечных заболеваний.
Миопатии
В ниже приведенной таблице представлены наиболее часто встречающиеся формы нервно-мышечных заболеваний.
Миопатии

II. Неврогенные амиотрофии.
А. Спинальные формы:
а) детская амиотрофия Верднига – Гофмана;
II. Неврогенные амиотрофии.
А. Спинальные формы:
а) детская амиотрофия Верднига – Гофмана;

Патогенез первично-прогрессирующих мышечных дистрофий – миопатий
Можно считать уже доказанным, что у
Патогенез первично-прогрессирующих мышечных дистрофий – миопатий
Можно считать уже доказанным, что у

У больных миопатией отмечается усиленное выделение с мочой таких аминокислот как
У больных миопатией отмечается усиленное выделение с мочой таких аминокислот как
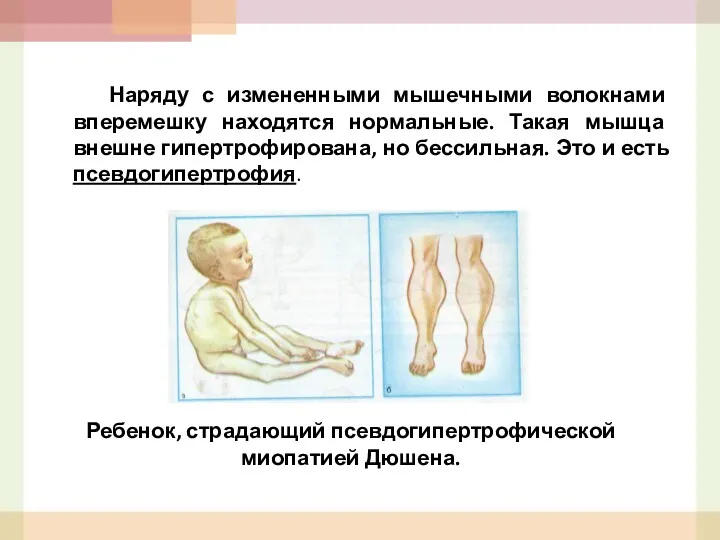
Наряду с измененными мышечными волокнами вперемешку находятся нормальные. Такая мышца внешне
Наряду с измененными мышечными волокнами вперемешку находятся нормальные. Такая мышца внешне
Клиника миопатий связана с прогрессирующей мышечной атрофией и нарастающей слабостью, как
Клиника миопатий связана с прогрессирующей мышечной атрофией и нарастающей слабостью, как
Постепенно увеличивается лордоз грудного отдела позвоночника (поза верблюда), появляются осиная талия,
Постепенно увеличивается лордоз грудного отдела позвоночника (поза верблюда), появляются осиная талия,
У этих больных полностью выпадают сухожильные рефлексы, объем активных движений постепенно
У этих больных полностью выпадают сухожильные рефлексы, объем активных движений постепенно

Рассмотрим кратко ряд клинических форм заболеваний:
а) Псевдогипертрофическая амиотрофия Дюшена.
Болезнь начинает проявляться
Рассмотрим кратко ряд клинических форм заболеваний:
а) Псевдогипертрофическая амиотрофия Дюшена.
Болезнь начинает проявляться
б) Детская спинальная амиотрофия Верднига – Гофмана.
Болезнь начинается с первых месяцев
б) Детская спинальная амиотрофия Верднига – Гофмана.
Болезнь начинается с первых месяцев
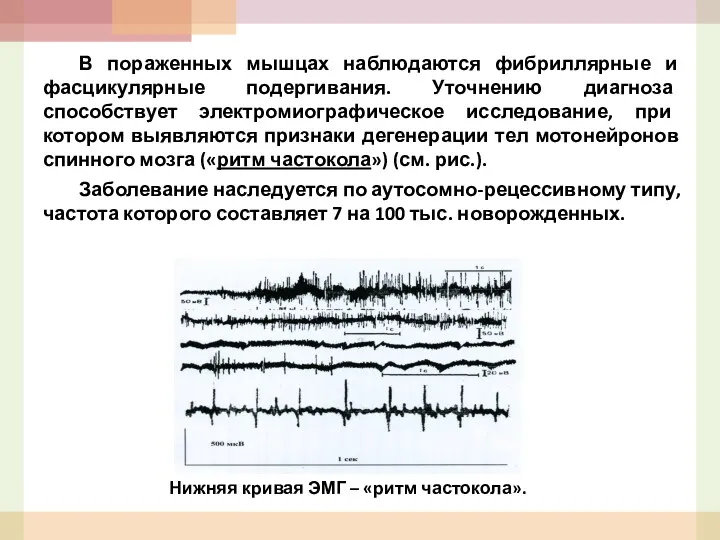
В пораженных мышцах наблюдаются фибриллярные и фасцикулярные подергивания. Уточнению диагноза способствует
В пораженных мышцах наблюдаются фибриллярные и фасцикулярные подергивания. Уточнению диагноза способствует
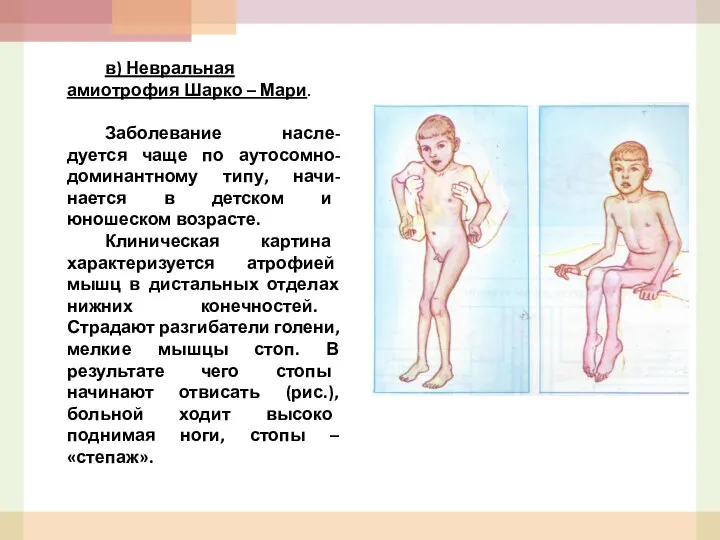
в) Невральная амиотрофия Шарко – Мари.
Заболевание насле-дуется чаще по аутосомно-доминантному типу,
в) Невральная амиотрофия Шарко – Мари.
Заболевание насле-дуется чаще по аутосомно-доминантному типу,

Наряду с двигательными нарушениями для этих больных характерны чувствительные расстройства по
Наряду с двигательными нарушениями для этих больных характерны чувствительные расстройства по

г) Миастения.
Основным симптомом заболевания является нарастающая мышечная слабость, резко усиливающаяся при
г) Миастения.
Основным симптомом заболевания является нарастающая мышечная слабость, резко усиливающаяся при

Специальные исследования показали нарушение синтеза ацетилхолина при избытке холинэстеразы в тканях
Специальные исследования показали нарушение синтеза ацетилхолина при избытке холинэстеразы в тканях

При миастенических кризах (резкое нарастание симптомов заболевания) рекомендуется срочное введение 0,5
При миастенических кризах (резкое нарастание симптомов заболевания) рекомендуется срочное введение 0,5
 Современная лабораторная диагностика туберкулеза. Опережая время
Современная лабораторная диагностика туберкулеза. Опережая время Күйік алған науқастарды тағами тұрғыдан қолдау
Күйік алған науқастарды тағами тұрғыдан қолдау Токсикомания. Синдром психической зависимости
Токсикомания. Синдром психической зависимости Практическое занятие №2: Клиническая классификация туберкулеза. Клинические формы туберкулеза органов дыхания
Практическое занятие №2: Клиническая классификация туберкулеза. Клинические формы туберкулеза органов дыхания Дыхательная гимнастика для школьников по методике Стрельниковой А.Н
Дыхательная гимнастика для школьников по методике Стрельниковой А.Н Синдромы и методы функциональной диагностики при патологии ЖВП и печени
Синдромы и методы функциональной диагностики при патологии ЖВП и печени Kombis etiologiuri klassifikaciya
Kombis etiologiuri klassifikaciya Оказание первой помощи при несчастных случаях на производстве
Оказание первой помощи при несчастных случаях на производстве Массаж для лошадей. Применение и правила выполнения
Массаж для лошадей. Применение и правила выполнения Современные хирургические методы лечения
Современные хирургические методы лечения Глазная биосовместимость
Глазная биосовместимость Астматический статус
Астматический статус Этиология және патогенез
Этиология және патогенез Особенности онкологических заболеваний у детей
Особенности онкологических заболеваний у детей Здоровье школьника – правильная осанка
Здоровье школьника – правильная осанка Сестринская помощь при осуществлении диагностических мероприятий пациентам инфекционного профиля. (Лекция 3)
Сестринская помощь при осуществлении диагностических мероприятий пациентам инфекционного профиля. (Лекция 3) Жас нәрестелердің пневмониясы
Жас нәрестелердің пневмониясы Недоношені діти
Недоношені діти Ортомиксовирусы, вирусы гриппа и ОРЗ
Ортомиксовирусы, вирусы гриппа и ОРЗ Инфаркт миокарда
Инфаркт миокарда Балалардың уақытша және тұрақты тістерін жұлу ерекшеліктері
Балалардың уақытша және тұрақты тістерін жұлу ерекшеліктері Гальванизация - лечение постоянным током низкого напряжения
Гальванизация - лечение постоянным током низкого напряжения Заболевание половых органов у девочек и их профилактика. Вульвагинит
Заболевание половых органов у девочек и их профилактика. Вульвагинит Проблема вкусовой привлекательности рациона при ХБП и способы ее повышения
Проблема вкусовой привлекательности рациона при ХБП и способы ее повышения Кишечный шов
Кишечный шов Диагностика плеврита, ХОБЛ, легочного сердца
Диагностика плеврита, ХОБЛ, легочного сердца Алынбайтын протездер
Алынбайтын протездер Bronchitis in children
Bronchitis in children