Наследственные болезни накопления: Мукополисахаридозы. Муковисцидоз. Причина, патогенез, клиника, диагностика, лечение,прогноз презентация
- Наследственные болезни накопления: Мукополисахаридозы. Муковисцидоз. Причина, патогенез, клиника, диагностика, лечение,прогноз

Содержание
- 2. Мукополисахаридозы (МПС) Мукополисахаридозы (МПС) -группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена гликозаминогликанов (ГАГ), проявляющихся
- 3. Различают следующие основные типы мукополисахаридозов: МПС I типа: Н типа – синдром Гурлер, H-S - синдром
- 4. Мукополисахаридоз типа I-Н (синдром Гурлер). Впервые описан немецким педиатром Гурлер (G. Hurler) в 1919 г. Часто
- 5. Признаки болезни появляются уже на первом году жизни, а к 1—2 годам все клинические проявления достаточно
- 7. ДИФ диагноз В начале развития болезни у детей раннего возраста синдром Гурлер необходимо дифференцировать с врожденными
- 8. Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер). Впервые описан американским офтальмологом Шейе (Н.G. Scheie) в
- 9. При рождении признаки болезни отсутствуют. Первые симптомы (ограничение разгибания пальцев рук) появляются в 3—6 лет. В.к,
- 11. Мукополисахаридоз типа II (синдром Гунтера). Клинические симптомы появляются позднее, чем при синдроме Гурлер (у детей старше
- 12. Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия. Синдром Гунтера у мальчика 2
- 14. Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо). Описан американским педиатром Санфилиппо (S.J. Sanfilippo) в 1963 г.
- 16. Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио). Заболевание в 1929 г. независимо друг от друга впервые
- 17. резкая задержка роста (рост взрослого больного около 100 см), непропорциональное телосложение (относительно короткое туловище, микроцефалия, короткая
- 18. Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами) в 1960 г. впервые описан
- 19. Отмечаются сгибательные контрактуры суставов верхних конечностей (больные не могут поднять руки вверх); с возрастом появляются контрактуры
- 20. Мукополисахаридоз типа VII (синдром Слая). Описан Слаем (W.S. Sly) в 1973 г. Клинические проявления схожи с
- 21. Мукополисахаридоз типа VIII (синдром Ди Ферранте). Описан Ди Ферранте (N. Di Ferrante) и др. в 1978
- 22. Жалобы на: отставание в росте; грубые черты лица увеличение размеров головы; деформация скелета; скованность в суставах;
- 23. Лабораторные исследования: ОАК: анемия, лейкопения, у 50% больных в лейкоцитах можно обнаружить зернистость Альдера. Биохимический анализ
- 24. Дифференциальная диагностика Синдром Гурлера – 1 тип Синдром Шейе – 1 тип S Синдром Хантера –
- 25. Дифференциальный диагноз:
- 27. Лечение : Режим в зависимости от тяжести и формы заболевания: от общего до постельного. Диета стол
- 28. При судорожном синдроме: · карбамазепин 0,2гр таблетках 20мг/кг 1 раз в день курс в зависимости от
- 29. По показаниям при наличии железодефицитной анемии: · актиферрин р-р 30мл – по 5 мл 1-2 раза
- 30. Другие виды лечения, оказываемые на амбулаторном уровне: · индивидуальные занятия с логопедом; · занятия с психологом;
- 31. Что такое Муковисцидоз у детей Название заболевания происходит от латинских слов mucus «слизь» и viscidus «вязкий».
- 32. Патогенез (что происходит?) во время Муковисцидоза у детей: Секрет экзокринных желёз становится особенно вязким, что вызывает
- 33. Различают следующие формы муковисцидоза: Лёгочная – возникает вследствие изменения состава слизи и её застоя в лёгких,
- 34. Легочная Железы слизистой оболочки дыхательных путей производят большое количество секрета, который из-за повышенной вязкости забивает мелкие
- 35. Признаком лёгочной формы муковисцидоза являются деформированные пальцы (симптом барабанных палочек)
- 36. http://vseroditelyam.ru/mukoviscidoz-u-detej/
- 37. Кишечная Нарушения со стороны ЖКТ обусловливаются секреторной недостаточностью многих органов пищеварительной системы. Например, вязкость секрета вызывает
- 39. Диагностические критерии Жалобы и анамнез: повторные и рецидивирующие пневмонии с затяжным течением, хронические изменения со стороны
- 40. Физикальное обследование: при классической картине MB больные дети имеют характерный внешний вид: «кукольное» лицо; расширенная, деформированная
- 41. Инструментальные исследования Рентген-пленочный тест: полное отсутствие переваривания желатины или ее разведения в разведениях 1: 20, 1:
- 43. Скачать презентацию
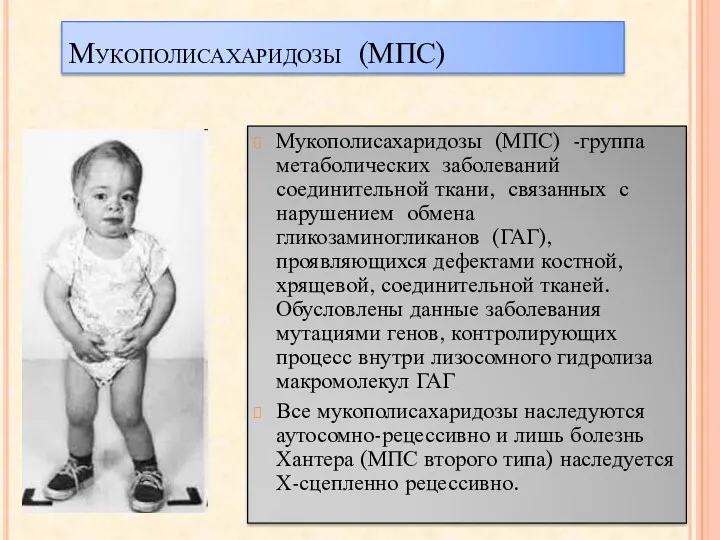
Мукополисахаридозы (МПС)
Мукополисахаридозы (МПС) -группа метаболических заболеваний соединительной ткани, связанных с
Мукополисахаридозы (МПС)
Мукополисахаридозы (МПС) -группа метаболических заболеваний соединительной ткани, связанных с

Различают следующие основные типы мукополисахаридозов:
МПС I типа: Н типа – синдром
Различают следующие основные типы мукополисахаридозов:
МПС I типа: Н типа – синдром
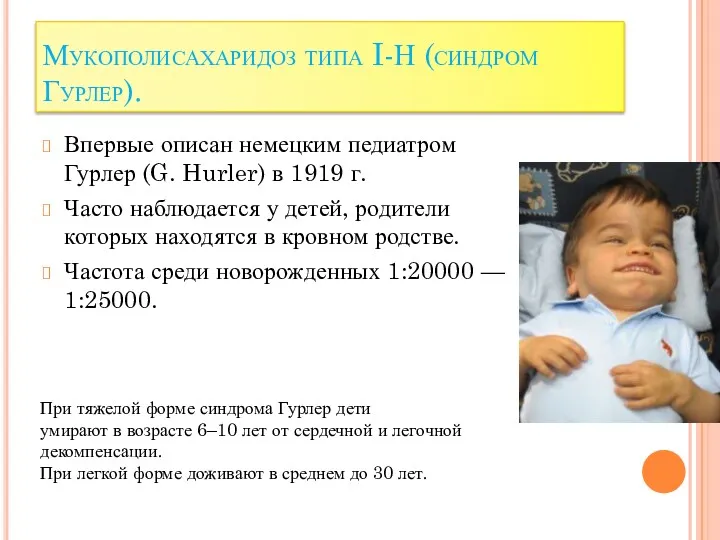
Мукополисахаридоз типа I-Н (синдром Гурлер).
Впервые описан немецким педиатром Гурлер (G.
Мукополисахаридоз типа I-Н (синдром Гурлер).
Впервые описан немецким педиатром Гурлер (G.
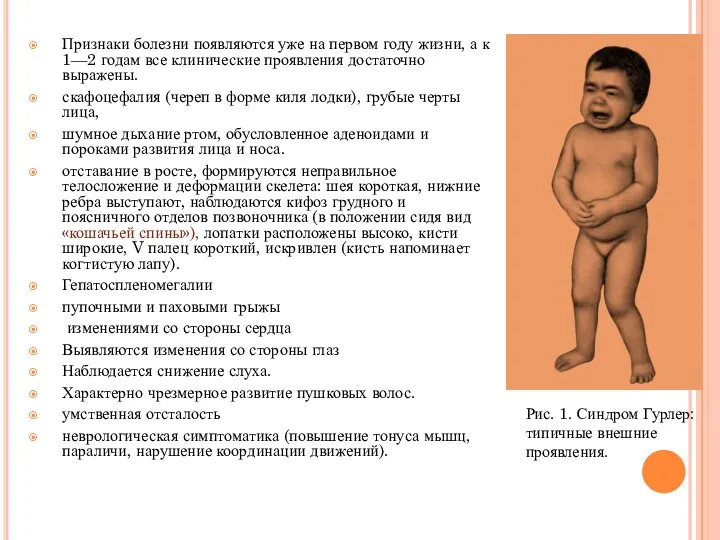
Признаки болезни появляются уже на первом году жизни, а к 1—2
Признаки болезни появляются уже на первом году жизни, а к 1—2


ДИФ диагноз
В начале развития болезни у детей раннего возраста синдром Гурлер
ДИФ диагноз
В начале развития болезни у детей раннего возраста синдром Гурлер

Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер).
Впервые описан американским
Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер).
Впервые описан американским
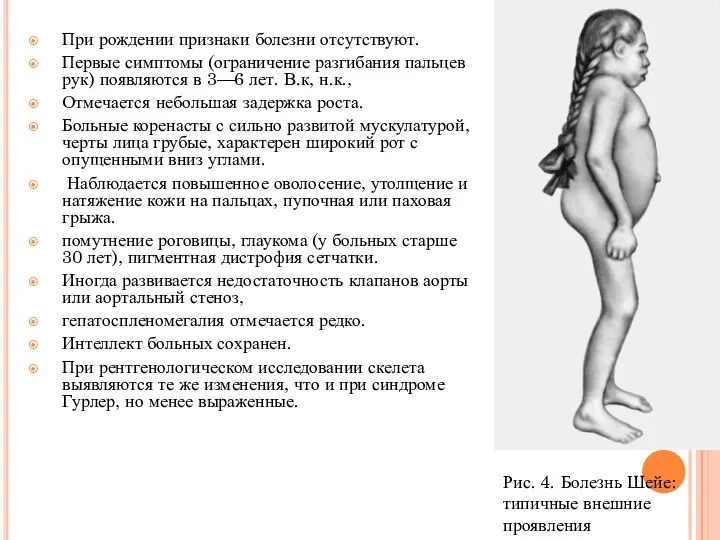
При рождении признаки болезни отсутствуют.
Первые симптомы (ограничение разгибания пальцев рук)
При рождении признаки болезни отсутствуют.
Первые симптомы (ограничение разгибания пальцев рук)


Мукополисахаридоз типа II (синдром Гунтера).
Клинические симптомы появляются позднее, чем при
Мукополисахаридоз типа II (синдром Гунтера).
Клинические симптомы появляются позднее, чем при
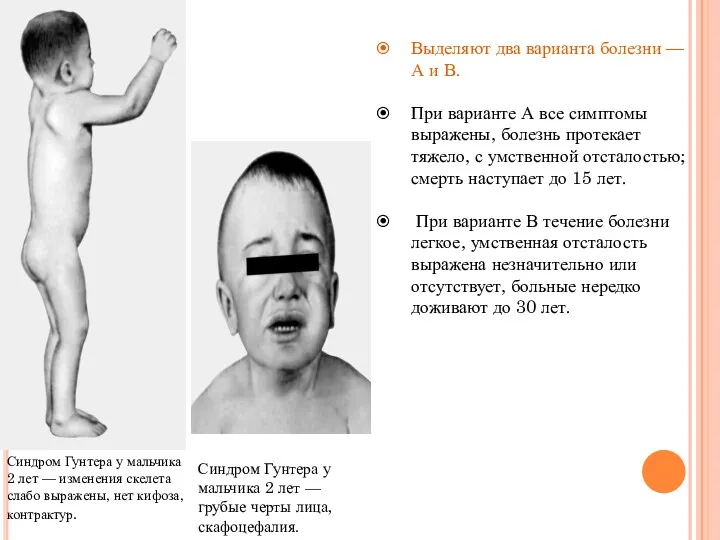
Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия.
Синдром
Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия.
Синдром


Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо).
Описан американским педиатром Санфилиппо
Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо).
Описан американским педиатром Санфилиппо

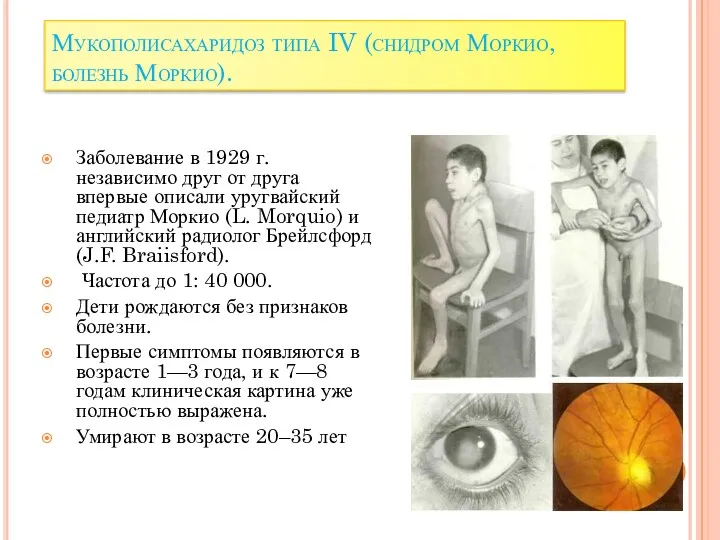
Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио).
Заболевание в 1929 г.
Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио).
Заболевание в 1929 г.
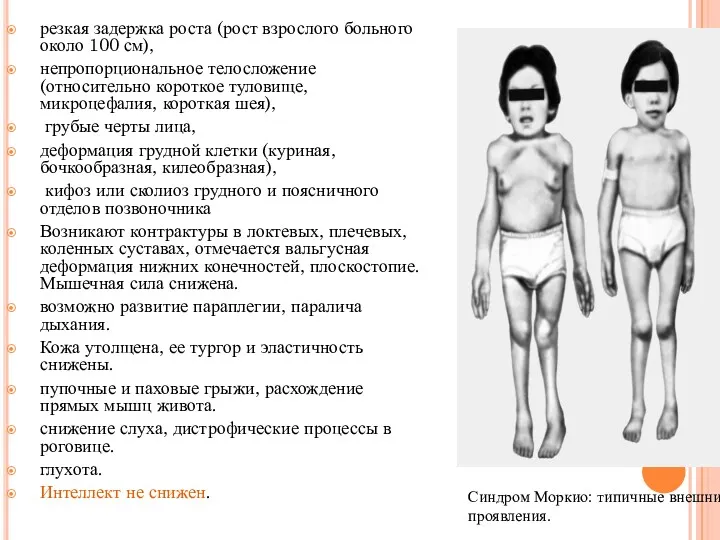
резкая задержка роста (рост взрослого больного около 100 см),
непропорциональное телосложение
резкая задержка роста (рост взрослого больного около 100 см),
непропорциональное телосложение
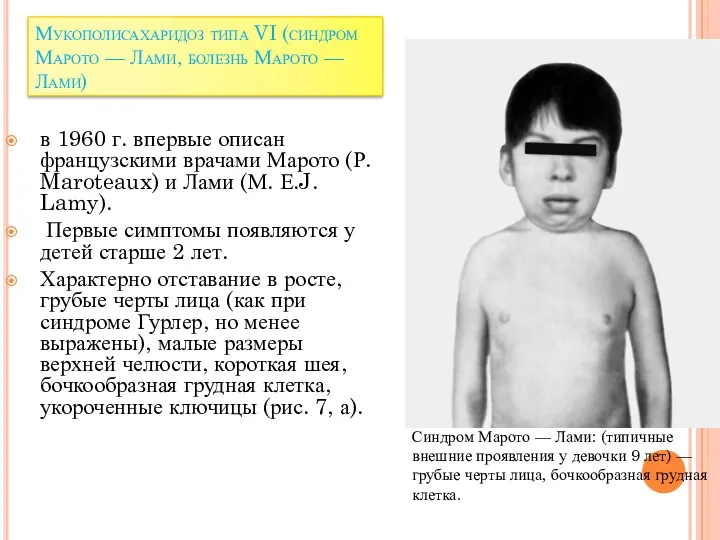
Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами)
Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами)
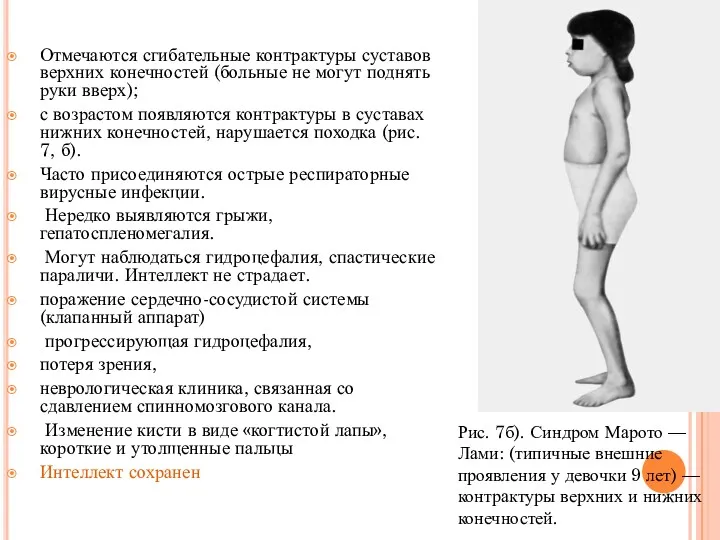
Отмечаются сгибательные контрактуры суставов верхних конечностей (больные не могут поднять руки
Отмечаются сгибательные контрактуры суставов верхних конечностей (больные не могут поднять руки

Мукополисахаридоз типа VII (синдром Слая).
Описан Слаем (W.S. Sly) в 1973
Мукополисахаридоз типа VII (синдром Слая).
Описан Слаем (W.S. Sly) в 1973

Мукополисахаридоз типа VIII (синдром Ди Ферранте).
Описан Ди Ферранте (N. Di
Мукополисахаридоз типа VIII (синдром Ди Ферранте).
Описан Ди Ферранте (N. Di

Жалобы на:
отставание в росте;
грубые черты лица
увеличение размеров головы;
деформация
Жалобы на:
отставание в росте;
грубые черты лица
увеличение размеров головы;
деформация

Лабораторные исследования:
ОАК: анемия, лейкопения, у 50% больных в лейкоцитах можно обнаружить зернистость
Лабораторные исследования:
ОАК: анемия, лейкопения, у 50% больных в лейкоцитах можно обнаружить зернистость

Дифференциальная диагностика
Синдром Гурлера – 1 тип
Синдром Шейе – 1 тип S
Синдром
Дифференциальная диагностика
Синдром Гурлера – 1 тип
Синдром Шейе – 1 тип S
Синдром
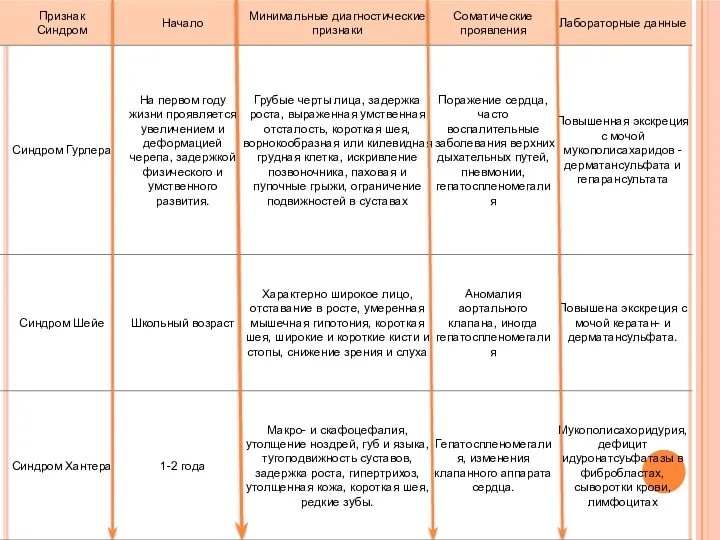
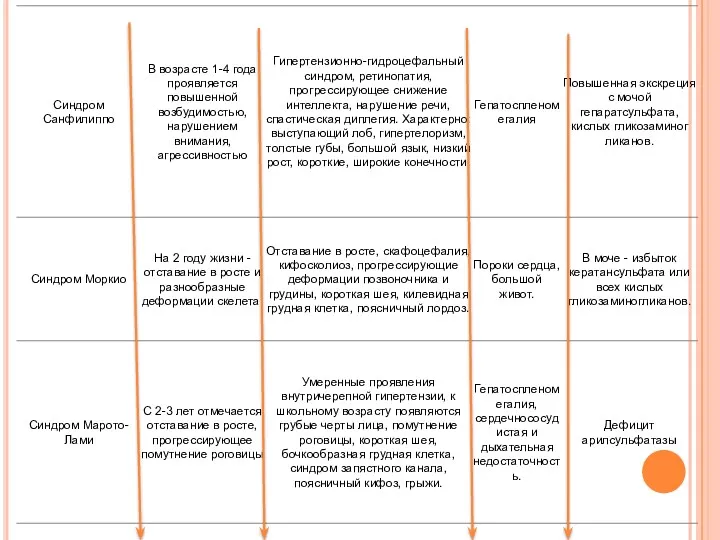
Дифференциальный диагноз:
Дифференциальный диагноз:

Лечение :
Режим в зависимости от тяжести и формы заболевания: от общего
Лечение :
Режим в зависимости от тяжести и формы заболевания: от общего

При судорожном синдроме:
· карбамазепин 0,2гр таблетках 20мг/кг 1 раз в день
При судорожном синдроме: · карбамазепин 0,2гр таблетках 20мг/кг 1 раз в день

По показаниям при наличии железодефицитной анемии:
· актиферрин р-р 30мл – по
По показаниям при наличии железодефицитной анемии: · актиферрин р-р 30мл – по

Другие виды лечения, оказываемые на амбулаторном уровне:
· индивидуальные занятия с логопедом;
·
Другие виды лечения, оказываемые на амбулаторном уровне:
· индивидуальные занятия с логопедом;
·
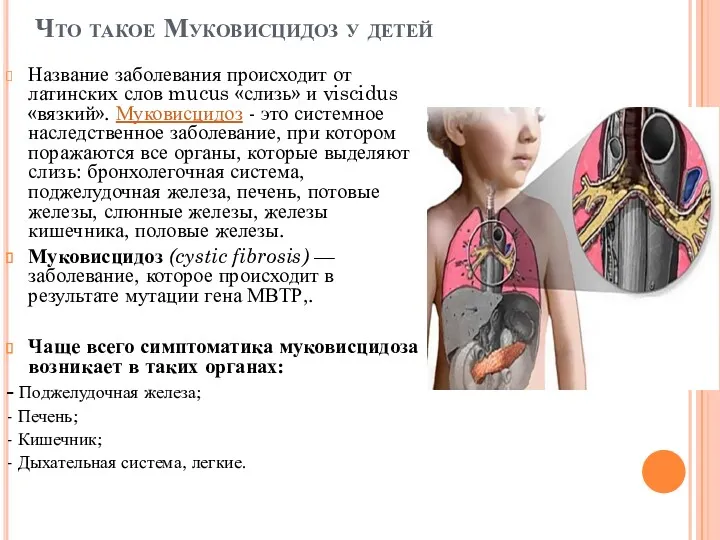
Что такое Муковисцидоз у детей
Название заболевания происходит от латинских слов mucus
Что такое Муковисцидоз у детей
Название заболевания происходит от латинских слов mucus
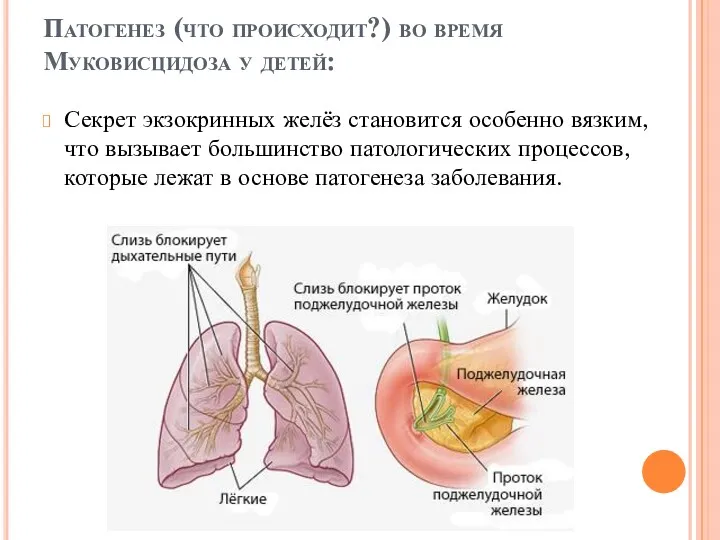
Патогенез (что происходит?) во время Муковисцидоза у детей:
Секрет экзокринных желёз становится
Патогенез (что происходит?) во время Муковисцидоза у детей:
Секрет экзокринных желёз становится

Различают следующие формы муковисцидоза:
Лёгочная – возникает вследствие изменения состава слизи и
Различают следующие формы муковисцидоза:
Лёгочная – возникает вследствие изменения состава слизи и

Легочная
Железы слизистой оболочки дыхательных путей производят большое количество секрета, который
Легочная
Железы слизистой оболочки дыхательных путей производят большое количество секрета, который
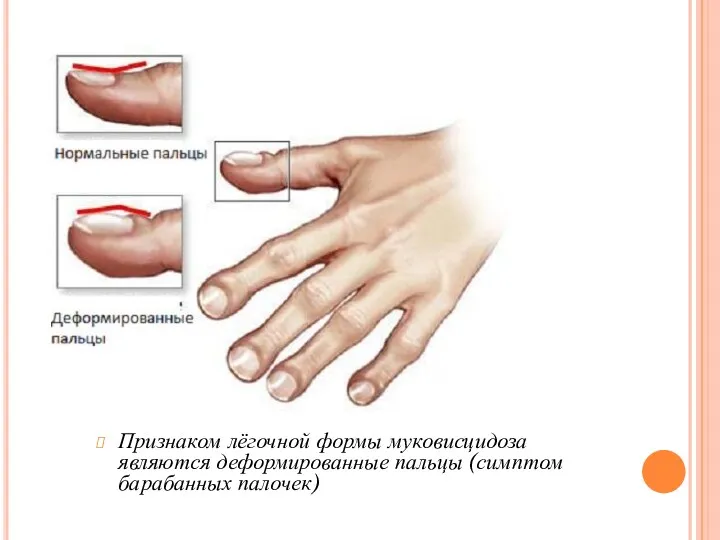
Признаком лёгочной формы муковисцидоза являются деформированные пальцы (симптом барабанных палочек)
Признаком лёгочной формы муковисцидоза являются деформированные пальцы (симптом барабанных палочек)

http://vseroditelyam.ru/mukoviscidoz-u-detej/
http://vseroditelyam.ru/mukoviscidoz-u-detej/

Кишечная
Нарушения со стороны ЖКТ обусловливаются секреторной недостаточностью многих органов пищеварительной
Кишечная
Нарушения со стороны ЖКТ обусловливаются секреторной недостаточностью многих органов пищеварительной


Диагностические критерии
Жалобы и анамнез: повторные и рецидивирующие пневмонии с затяжным течением, хронические
Диагностические критерии
Жалобы и анамнез: повторные и рецидивирующие пневмонии с затяжным течением, хронические

Физикальное обследование: при классической картине MB больные дети имеют характерный внешний вид:
Физикальное обследование: при классической картине MB больные дети имеют характерный внешний вид:

Инструментальные исследования
Рентген-пленочный тест: полное отсутствие переваривания желатины или ее разведения в
Инструментальные исследования Рентген-пленочный тест: полное отсутствие переваривания желатины или ее разведения в
 Организация медпомощи сельским жителям. Организация медпомощи работающим на промышленных предприятиях
Организация медпомощи сельским жителям. Организация медпомощи работающим на промышленных предприятиях Массаж при нарушениях обмена веществ. (Тема 5.8)
Массаж при нарушениях обмена веществ. (Тема 5.8) Хвороби дихальної системи
Хвороби дихальної системи Холера
Холера Медицинская эргономика и ее роль в сохранении здоровья
Медицинская эргономика и ее роль в сохранении здоровья Интенсивная терапия ОНМК, ЧМТ, судорожного синдрома
Интенсивная терапия ОНМК, ЧМТ, судорожного синдрома Методики рентгенологического исследования тонкой и толстой кишки
Методики рентгенологического исследования тонкой и толстой кишки Иммунитет. Иммунная система
Иммунитет. Иммунная система Вопросы инфекционной безопасности при работе с пациентами коронавирусной инфекцией COVID-19
Вопросы инфекционной безопасности при работе с пациентами коронавирусной инфекцией COVID-19 Внутрибольничные инфекции. Масштаб, проблемы и структура. Инфекционный процесс
Внутрибольничные инфекции. Масштаб, проблемы и структура. Инфекционный процесс Антидепрессанты (тимоаналептики)
Антидепрессанты (тимоаналептики) Топографическая анатомия и оперативная хирургия таза и промежности
Топографическая анатомия и оперативная хирургия таза и промежности Стоматологическое материаловедение. Классификация материалов
Стоматологическое материаловедение. Классификация материалов Физиология сенсорных систем. Анализаторы
Физиология сенсорных систем. Анализаторы Предоперационный период
Предоперационный период Дисциркуляторная энцефалопатия. Дифференциальная диагностика деменций
Дисциркуляторная энцефалопатия. Дифференциальная диагностика деменций Көк ірің таяқшасы
Көк ірің таяқшасы Невідкладні стани в ендокринології
Невідкладні стани в ендокринології OMS - Organizaţia Mondială a Sănătăţii
OMS - Organizaţia Mondială a Sănătăţii Болезнь Крона: современные аспекты диагностики и лечения
Болезнь Крона: современные аспекты диагностики и лечения Нарушение опорно-двигательного аппарата у детей
Нарушение опорно-двигательного аппарата у детей Позвоночник: причины искривления
Позвоночник: причины искривления Инфекционно-токсический шок при менингококковой инфекции. Мероприятия при ИТШ
Инфекционно-токсический шок при менингококковой инфекции. Мероприятия при ИТШ Современные клещевые инфекции
Современные клещевые инфекции Заболевания мочевой системы. Занятие 6
Заболевания мочевой системы. Занятие 6 Ревматическая лихорадка
Ревматическая лихорадка лекция №3,4 (4 сем) Эндодонтическое лечение зубов 03.03.20 — копия 2
лекция №3,4 (4 сем) Эндодонтическое лечение зубов 03.03.20 — копия 2 Влияние полноценного питания на организм
Влияние полноценного питания на организм