- Наследственные болезни обмена веществ

Содержание
- 2. Включают более 700 нозологических форм. Большинство относится к орфанным заболеваниям, т.к. их распространенность составляет менее 10
- 3. Актуальность 80% НБО манифестируют в детском возрасте. Эти болезни связаны с нарушением определенного метаболического пути. Биохимические
- 4. Наиболее распространены и изучены: нарушение обмена аминокислот / органических кислот, нарушение обмена углеводов, нарушение обмена липидов.
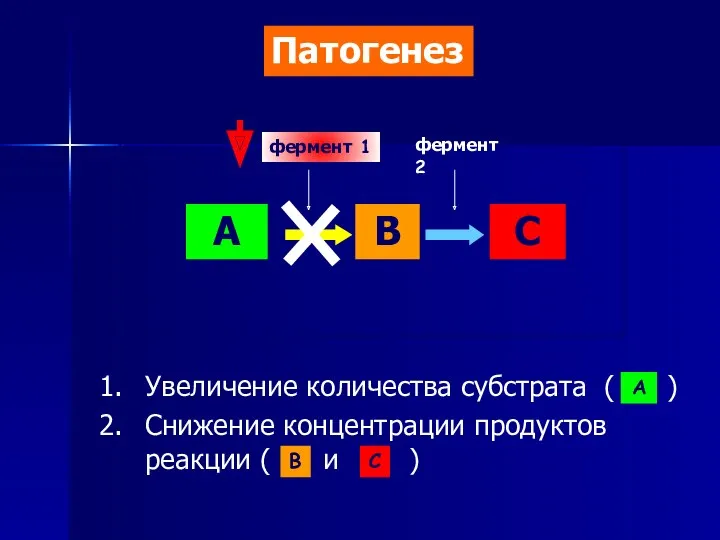
- 5. А В С фермент 1 фермент 2 Увеличение количества субстрата ( ) Снижение концентрации продуктов реакции
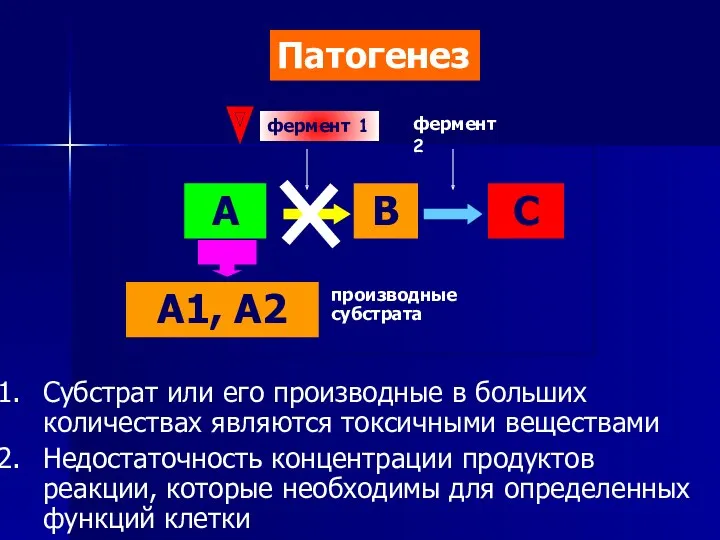
- 6. А В С А1, А2 фермент 1 фермент 2 Субстрат или его производные в больших количествах
- 7. Диагностика С целью раннего выявления НБО применяют программы массового и селективного скрининга. Новая эра скрининга –
- 8. АМИНОКИСЛОТ Наследственные болезни обмена
- 9. Фенилкетонурия (ФКУ) фенилпировиноградная олигофрения, болезнь Феллинга наследственное заболевание аминокислотного обмена, в основе которого лежит нарушение превращения
- 10. Фенилкетонурия Самое частое наследственное нарушение обмена аминокислот. Частота ФКУ 1:8.000, у россиян 1:4.000 –6.500, при УМО-
- 11. Фенилаланин является незаменимой аминокислотой для построения белковой молекулы, являясь предшественником тироксина, адреналина и меланина Отсутствие фенилаланин-4-гидроксилазы
- 12. Патогенез Фенилпировиноградная кислота является нейротропным ядом, и приводит к повышению возбудимости мышц, гиперефлексии, тремору, судорогам, УМО,
- 13. 1.Задержка психического развития. 2.Судорожный синдром. 3.Склонность к развитию дерматита (выделение аномальных метаболитов кожными железами). 4.Нарушение пигментного
- 14. светлые волосы, светло-голубые глаза, светлая кожа Нарушение пигментного обмена с рождения:
- 15. Неврологический статус больных ФКУ Рано изменяется мышечный тонус – от гипотонии до ригидности; при стоянии и
- 16. Первые 2-3 месяца жизни проходят без больших отклонений от нормы. К 4-6 месяцам появляется отставание в
- 17. Пациент с ФКУ
- 18. Диагностика фенилкетонурии Основной диагностический критерий - повышение концентрации ФА в крови. Нормальный уровень фенилаланина в крови
- 19. 2. положительная реакция Феллинга – выделение с мочой повышенных количеств фенилпировиноградной кислоты и других кетоновых кислот,
- 20. Установить диагноз в доклинической стадии, не позднее 2-го месяца жизни, когда могут проявиться первые признаки болезни.
- 21. Скрининг на ФКУ Проводится всем новорожденным на 4-5 день жизни. Для определения концентрации ФА в сухом
- 22. Диетотерапия Основной принцип лечения – ограничение поступления ФА с пищей. Диета обычно отменяется к 6-10-летнему возрасту.
- 23. Запрещено: мясо, колбасы, рыба, творог, сыр, яйца, соя, орехи, хлеб, крупы, мучное, шоколад, фасоль, горох, желатин
- 24. Лечение ФКУ Если лечение ФКУ начато со 2-й недели жизни, то интеллект остается сохранным в 90%
- 25. Наследственные болезни обмена УГЛЕВОДОВ
- 26. Галактоземия Заболевание относится к нарушениям обмена простых сахаров. Возникает при вскармливании молоком при наследственной непереносимости лактозы,
- 27. Патогенез галактоземии - накопление галактозы и галактозо-1-фосфата в крови и тканях Выделяют 3 формы, вызванных недостаточностью
- 28. Приводит к накоплению галактозы в крови, головном мозге, печени, почках и ЖКТ. Классическая форма галактоземии Отсутствие
- 29. Острое начало на 1-м месяце жизни при кормлении молоком: Неукротимая рвота, понос, гепатоспленомегалия, желтуха; аминоацидурия и
- 30. Общий анализ крови -анемия. Биохимия крови: Повышение уровня: галактозы , АСТ, АЛТ; билирубина. Снижение глюкозы, общего
- 31. Лечение галактоземии Диета с исключением галактозы. Замена грудного и коровьего молока, молочных продуктов смесями с соевым
- 32. Фруктозурия А/р заболевание с частотой 0,8:100.000. В основе - недостаточное количество фруктозо-1-фосфат-альдолазы, который переводит фруктозу в
- 33. В зависимости от степени недостатка ферментов выделяют легкие и тяжелые формы. Легкие формы – симптомы незначительные,
- 34. Клиника Гипогликемические состояния после приема пищи, содержащей фруктозу; увеличении печени с биохимическими признаками нарушения липидного обмена,
- 35. Диагностика Проба с нагрузкой фруктозой вызывает гипогликемическую реакцию. Условием ее проведения должна быть готовность к борьбе
- 36. Лечение Диета с исключением из рациона ребенка «вредных» продуктов питания, не менее трех лет. Запрещенные продукты:
- 37. Наследственные болезни обмена ЛИПИДОВ К этим заболеваниям относятся липидозы и лейкодистрофии
- 38. НБО липидов
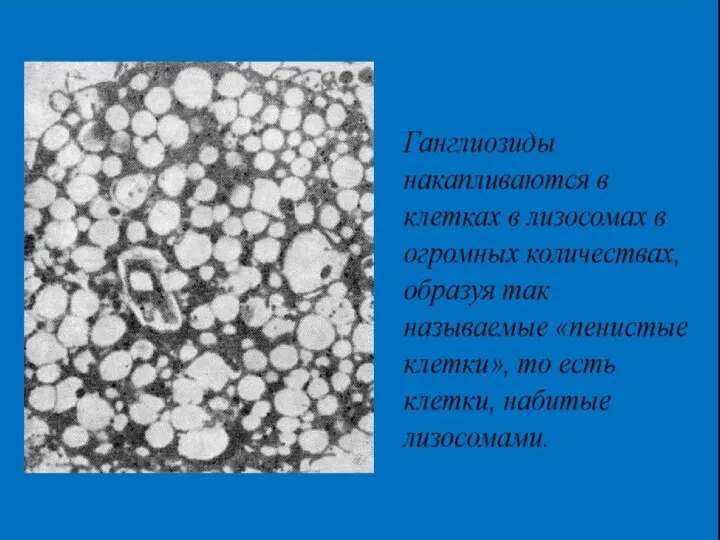
- 39. Амавротическая идиотия — редкое наследственное заболевание с АР типом наследования, при котором в тканях накапливаются ганглиозиды
- 40. Частота встречаемости заболевания: 1:300.000 (среди евреев ашкинази– 1:3600). Около 3% населения являются носителями болезни. Ген локализован
- 41. Патогенез болезни Тея — Сакса генетический дефект гена, приводит к отсутствию фермента гексозоаминидазы A, находящегося в
- 44. Выделяют 4 формы в зависимости от времени начала заболевания и клиники. Наиболее изучена инфантильная форма –
- 45. Клиника болезни Тея — Сакса 1. Задержка, а затем регресс развития (после инфекций, травм): утрата приобретенных
- 46. 5. Фармакорезистентные судороги 6. Центральные тетрапарезы. 7. МРТ головного мозга: атрофия нейронов коры, клеток Пуркинье, ядер
- 47. Характерный диагностический признак синдром «вишневой косточки», возникающий из-за атрофии ганглиозных клеток сетчатки и просвечивания сосудистой оболочки
- 48. Диагностика, лечение, прогноз В плазме крови: увеличено содержание цереброзидов и сульфатидов; В Er увеличение свободного холестерина
- 49. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ лизосомные болезни накопления
- 50. Это органеллы, в матриксе которых содержатся различные ферменты, которые осуществляют внутриклеточное переваривание различных химических соединений и
- 51. Лизосомные болезни накопления (ЛБН) Обусловлены мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул
- 52. Лизосомные болезни накопления(ЛБН) Число известных форм – около 50 Наследуются по аутосомно-рецессивному типу (кроме болезни Фабри
- 53. Нормальный катаболизм Субстрат Нарушение активности фермента Накопление субстрата блокированной реакции Патогенез ЛБН
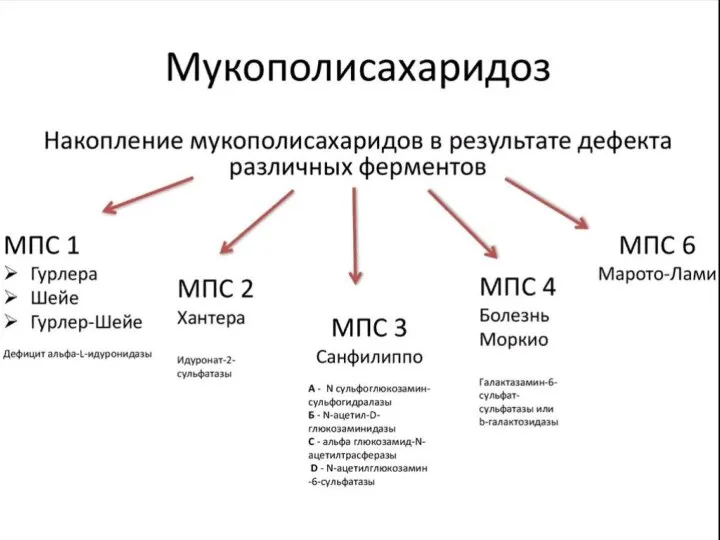
- 54. Классификация ЛБН В зависимости от накапливаемых субстратов: Сфинголипидозы (болезнь Гоше, метахроматическая лейкодистрофия, болезнь Краббе) Ганглиозидозы (болезнь
- 55. Лизосомальные болезни обмена
- 56. Клинические проявления ЛБН Прогрессирующий характер заболевания Наличие различного по продолжительности интервала нормального развития (от нескольких месяцев
- 57. МУКОПОЛИСАХАРИДОЗЫ
- 58. Мукополисахаридозы группа ЛБН, связанных с нарушением обмена гликозаминогликанов ГАГ являются важнейшими компонентами основного структурного белка волос
- 60. Патогенез МПС I типа Недостаточность фермента α-L-идуронидазы Нарушение отщепления терминального остатка идуроновой кислоты Нарушение внутрилизосомной деградации
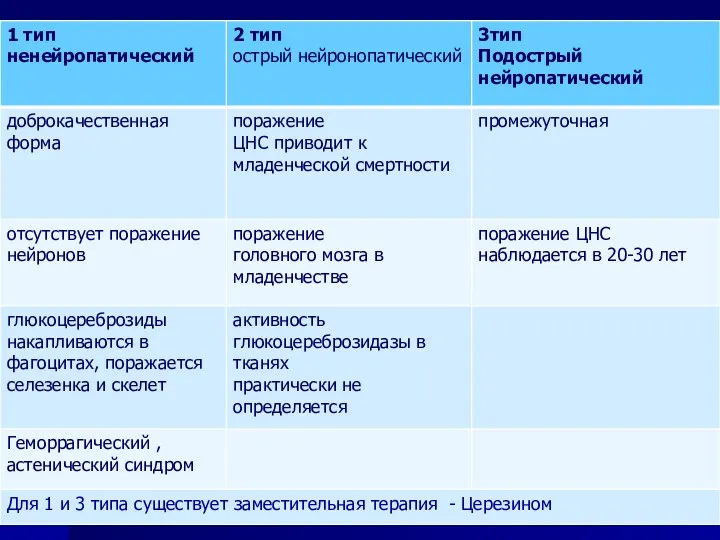
- 61. МПС, I тип. В зависимости от клинической картины выделяют 3 клинические варианта: Синдром Гурлер Шейе Гурлер-Шейе
- 62. МПС тип I (синдром Гурлер) Эта форма встречается чаще остальных и была описана ранее других синдромов.
- 63. gargoille – лепные изображения на фронтонах готических соборов Впервые данная форма была описана в 1919 году
- 64. Симптомы проявляются с рождения, к 1-2годам клиническая картина выражена полностью. Характерна мультисистемность поражения: МПС тип I
- 65. К концу 1 года жизни становятся очевидными изменение черт лица Макроцефалия, увеличение лобных бугров, широкий нос
- 66. Тугоподвижность и контрактуры тазобедренных и коленных суставов Плечевых и локтевых суставов Тугоподвижность суставов
- 67. Ребенок с I типом МПС – синдром Гурлер Генотип девочки: Q70X/Q70X
- 68. Больные больше похожи друг на друга, чем на своих здоровых братьев и сестер
- 69. Синдром Гурлер манифестирует на 1 году жизни тяжелая соматическая и неврологическая патология Синдром Гурлер—Шейе манифестирует в
- 70. Дети 3,5 и 5 лет с синдромами Гурлер (слева) и Гурлер-Шейе (справа)
- 71. Диагностика МПС характерные клинические проявления, определении экскреции с мочой ГАГ пренатальная диагностика (определение активности лизосомальных ферментов
- 72. Фермент-заместительная терапия : альдуразим при МПС 1 типа, элапраза при МПС 2 типа, наглазим при 6
- 73. Болезнь Ниманна-Пика Редкое наследственное аутосомно-рецессивное заболевание. Частота 1:10.000. Среди евреев-ашкинази 1:100 Мутации гена сфингомиелин-фосфодиэстеразы I приводит
- 74. А и Б - дефицит сфингомиелиназы С – нарушение транспорта липидов
- 75. Болезнь Ниманна-Пика А Ранняя манифестация заболевания Гепатоспленомегалия Прогрессирующая церебеллярная атаксия Снижение интеллекта. Вертикальный надъядерный паралич взора!!
- 76. Активность сфингомиелиназы – 0, 03 нмоль/мг/час (норма 7,5 – 60,0 нмоль/мг/час)
- 77. Болезнь Гоше Мутация в гене GBA приводит к дефициту фермента- глюкоцереброзидазы, в результате в лизосомах органах
- 78. Клетки Гоше
- 79. Клинически это проявляется: Гепатоспленомегалией Вследствие гиперплазии костного мозга развивается истончение костей, что приводит к их деформации
- 80. Клиника 3. Поражение костного мозга сопровождается панцитопенией. 4. Поражение ЦНС сопровождается судорогами, умственной деградацией
- 83. Профилактика лизосомных болезней накопления Пренатальная диагностика амниоцентез кордоцентез определение активности ферментов ДНК-диагностика
- 84. Основные термины медицинской генетики
- 85. Генетическая гетерогенность - разнообразие генетических причин наследственных болезней - феномен, когда клинически единое заболевание может быть
- 86. Причины генотипического полиморфизма: различные гены контролируют различные звенья одного и того же метаболического процесса, а поражение
- 87. Генотипический полиморфизм приводит к: многообразию отдельных симптомов; изменчивости этих признаков; появлению переходных и стертых форм; возникновению
- 88. Плейотропное действие – множественное действие гена, когда один ген влияет на развитие многих признаков 1 белок-фермент,
- 89. Плейотропное действие Плейотропное проявление мутантного гена, обнаруживается практически при всех наследственных заболеваниях. Серповидно-клеточная анемия. Замена одной
- 90. Пенетрантность генов - частота проявления того или иного гена, измеряемая частотой встречаемости признака в популяции Доминантный
- 91. Экспрессивность генов (выраженность проявления генетически детерминированного признака) Min Max Экспрессия генотипов может зависеть от внешних условий.
- 92. Экспрессивность генов Содержание хлора в поте у человека не более 40 ммоль/л, а при муковисцидозе (при
- 93. Гипотеза условного тропизма дефектных генов (Давыденков С.Н.) Мутантный патологический ген обнаруживает собственный плейотропизм независимый от остального
- 94. Микропризнаки или малые аномалии развития - это стойкий морфологический вариант изменения строения органа, при котором не
- 95. малые аномалии развития Не имеют серьезного медицинского или косметического значения, но часто выступают как значащие симптомы
- 96. Малые аномалии развития Диагностическая значимость микропризнаков увеличивается, если они сочетаются друг с другом. Любой микропризнак может
- 97. Малые аномалии развития обладают неравноценной диагностической значимостью Наибольшее значение имеют такие микропризнаки, которые встречаются редко и
- 98. Эпикант «Готическое» небо Синдактилия Гетерохромия
- 99. МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ Одна из первых медико-генетических консультаций в мире была создана отечественным невропатологом и генетиком С.Н.Давиденковым
- 100. - отрасль профилактической медицины, главной целью которой является снижение количества генетически обусловленных болезней и врожденных пороков
- 101. ЦЕЛЬ ГЕНЕТИЧЕСКОЙ КОНСУЛЬТАЦИИ установление степени генетического риска; помощь семье в принятии правильного решения по вопросам планирования
- 102. Информация о генетическом риске выдается в виде вероятностей, а степень риска определяется в форме процентов или
- 103. Генетический риск определяется двумя способами: Путем теоретических расчетов, основанных на генетических закономерностях; С помощью эмпирических данных.
- 104. ГЕНЕТИЧЕСКИЙ РИСК: Низкий (до 5%) – нет противопоказаний к деторождению в семье; Средний (6% до 20%)
- 105. ОСНОВНЫЕ СОСТАВЛЯЮЩИЕ МЕДИКО-ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ
- 106. ПОКАЗАНИЯ для медико-генетического консультирования: Наследственный дефект подозревается, и для уточнения диагноза требуются генетические методы исследования. Диагноз
- 107. Отставание ребенка в физическом или умственном развитии. Наличие МАР в сочетании с другими патологическими признаками: низкий
- 108. Кровное родство родителей больного ребенка. Повторные случаи мертворождения в семье при отсутствии акушерской патологии; возраст матери
- 109. Этапы медико-генетического консультирования Диагноз Прогноз Заключение Совет ВСЕ РЕШЕНИЯ ПО ПЛАНИРОВАНИЮ СЕМЬИ ПРИНИМАЮТСЯ ТОЛЬКО СУПРУГАМИ
- 110. Первичная профилактика наследственных болезней 1. Планирование деторождения Оптимальный возраст для женщины 21-35 лет; Отказ от деторождения
- 111. Опасность близкородственных браков У них родились 10 детей. 3 из которых умерли в возрасте до 10
- 112. Вторичная профилактика прерывание беременности при высоком риске наследственного заболевания, либо при пренатально диагностированной болезни
- 113. Третичная профилактика коррекция проявления патологических генотипов, т.е. нормокопирование при ФКУ, галактоземии и др.
- 115. Скачать презентацию

Включают более 700 нозологических форм.
Большинство относится к орфанным заболеваниям, т.к.
Включают более 700 нозологических форм.
Большинство относится к орфанным заболеваниям, т.к.

Актуальность
80% НБО манифестируют в детском возрасте.
Эти болезни связаны с нарушением определенного
Актуальность
80% НБО манифестируют в детском возрасте.
Эти болезни связаны с нарушением определенного

Наиболее распространены и изучены:
нарушение обмена аминокислот / органических кислот,
нарушение обмена
Наиболее распространены и изучены:
нарушение обмена аминокислот / органических кислот,
нарушение обмена
А
В
С
фермент 1
фермент 2
Увеличение количества субстрата ( )
Снижение концентрации продуктов
А
В
С
фермент 1
фермент 2
Увеличение количества субстрата ( )
Снижение концентрации продуктов
А
В
С
А1, А2
фермент 1
фермент 2
Субстрат или его производные в больших количествах
А
В
С
А1, А2
фермент 1
фермент 2
Субстрат или его производные в больших количествах

Диагностика
С целью раннего выявления НБО применяют программы массового и селективного скрининга.
Новая
Диагностика
С целью раннего выявления НБО применяют программы массового и селективного скрининга.
Новая

АМИНОКИСЛОТ
Наследственные болезни
обмена
АМИНОКИСЛОТ
Наследственные болезни
обмена
Фенилкетонурия (ФКУ)
фенилпировиноградная олигофрения,
болезнь Феллинга
наследственное заболевание аминокислотного обмена, в основе
Фенилкетонурия (ФКУ)
фенилпировиноградная олигофрения,
болезнь Феллинга
наследственное заболевание аминокислотного обмена, в основе

Фенилкетонурия
Самое частое наследственное нарушение обмена аминокислот.
Частота ФКУ 1:8.000,
у россиян
Фенилкетонурия
Самое частое наследственное нарушение обмена аминокислот.
Частота ФКУ 1:8.000,
у россиян
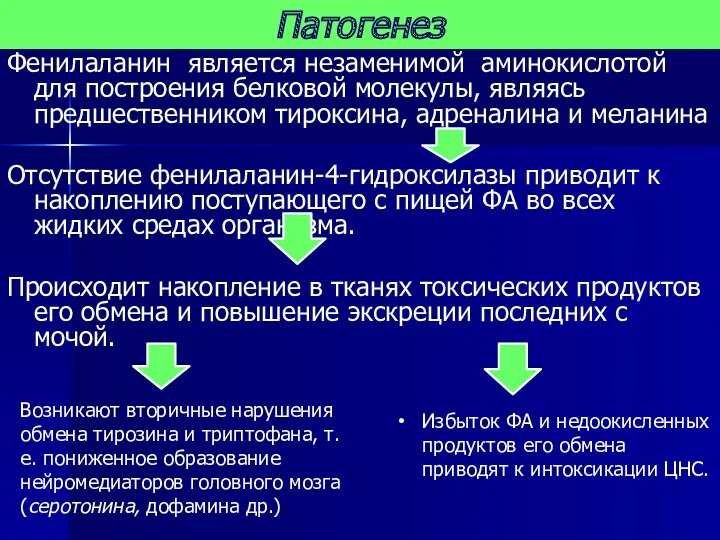
Фенилаланин является незаменимой аминокислотой для построения белковой молекулы, являясь предшественником тироксина,
Фенилаланин является незаменимой аминокислотой для построения белковой молекулы, являясь предшественником тироксина,
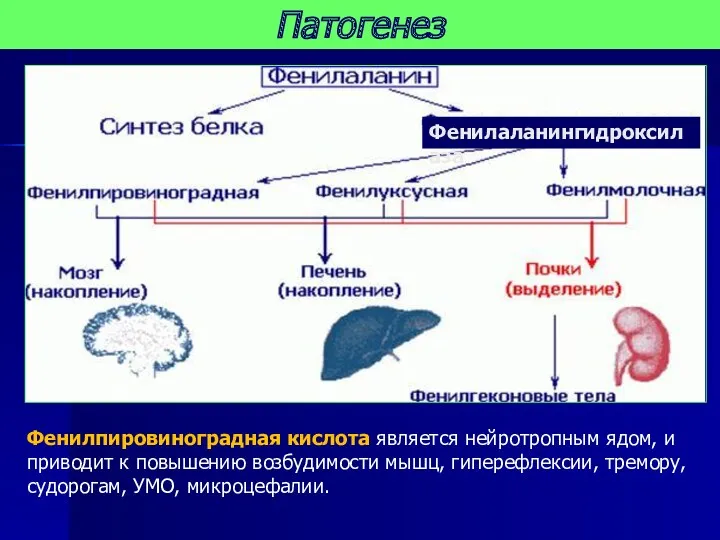
Патогенез
Фенилпировиноградная кислота является нейротропным ядом, и приводит к повышению возбудимости мышц,
Патогенез
Фенилпировиноградная кислота является нейротропным ядом, и приводит к повышению возбудимости мышц,

1.Задержка психического развития.
2.Судорожный синдром.
3.Склонность к развитию дерматита (выделение аномальных
2.Судорожный синдром.
3.Склонность к развитию дерматита (выделение аномальных

светлые волосы,
светло-голубые глаза,
светлая кожа
Нарушение пигментного обмена с рождения:
светлые волосы,
светло-голубые глаза,
светлая кожа
Нарушение пигментного обмена с рождения:
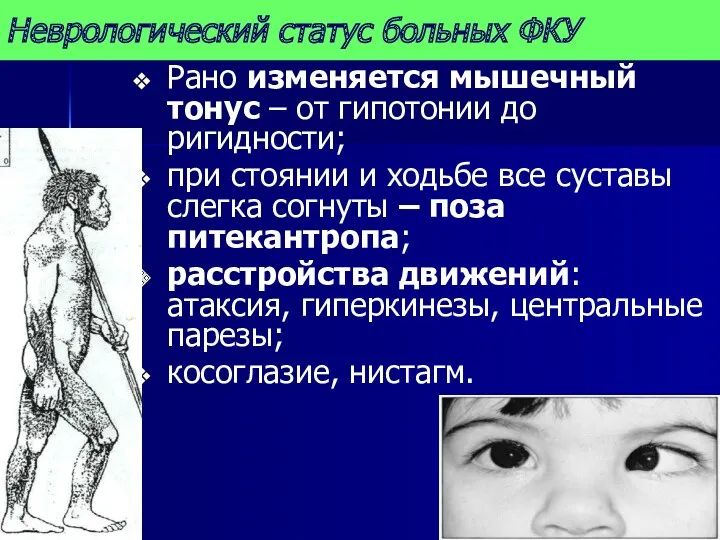
Неврологический статус больных ФКУ
Рано изменяется мышечный тонус – от гипотонии до
Неврологический статус больных ФКУ
Рано изменяется мышечный тонус – от гипотонии до

Первые 2-3 месяца жизни проходят без больших отклонений от нормы.
К 4-6
Первые 2-3 месяца жизни проходят без больших отклонений от нормы.
К 4-6
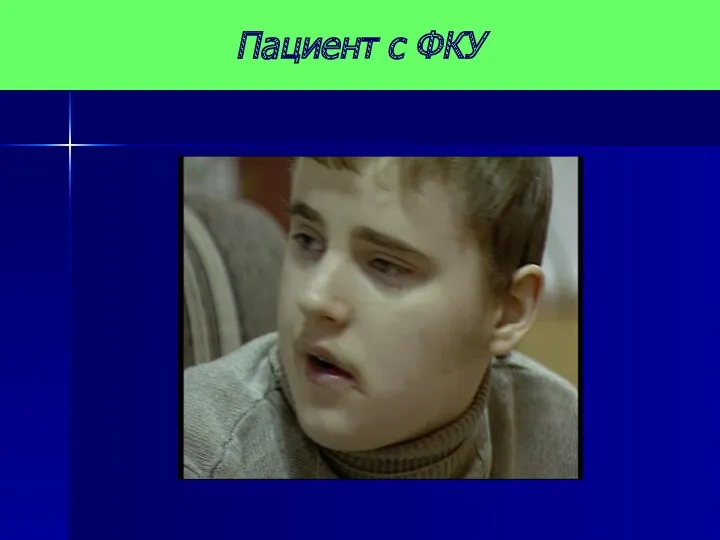
Пациент с ФКУ
Пациент с ФКУ

Диагностика фенилкетонурии
Основной диагностический критерий - повышение концентрации ФА в крови.
Нормальный уровень
Диагностика фенилкетонурии
Основной диагностический критерий - повышение концентрации ФА в крови.
Нормальный уровень

2. положительная реакция Феллинга
– выделение с мочой повышенных количеств фенилпировиноградной
2. положительная реакция Феллинга
– выделение с мочой повышенных количеств фенилпировиноградной
Установить диагноз в доклинической стадии, не позднее 2-го месяца жизни, когда
Установить диагноз в доклинической стадии, не позднее 2-го месяца жизни, когда

Скрининг на ФКУ
Проводится всем новорожденным на 4-5 день жизни. Для определения
Скрининг на ФКУ
Проводится всем новорожденным на 4-5 день жизни. Для определения

Диетотерапия
Основной принцип лечения –
ограничение поступления ФА с пищей.
Диета обычно отменяется
Диетотерапия
Основной принцип лечения –
ограничение поступления ФА с пищей.
Диета обычно отменяется

Запрещено: мясо, колбасы, рыба, творог, сыр, яйца, соя, орехи, хлеб, крупы,
Запрещено: мясо, колбасы, рыба, творог, сыр, яйца, соя, орехи, хлеб, крупы,

Лечение ФКУ
Если лечение ФКУ начато со 2-й недели жизни, то интеллект
Лечение ФКУ
Если лечение ФКУ начато со 2-й недели жизни, то интеллект

Наследственные болезни обмена
УГЛЕВОДОВ
Наследственные болезни обмена
УГЛЕВОДОВ

Галактоземия
Заболевание относится к нарушениям обмена простых сахаров.
Возникает при вскармливании
Галактоземия
Заболевание относится к нарушениям обмена простых сахаров.
Возникает при вскармливании
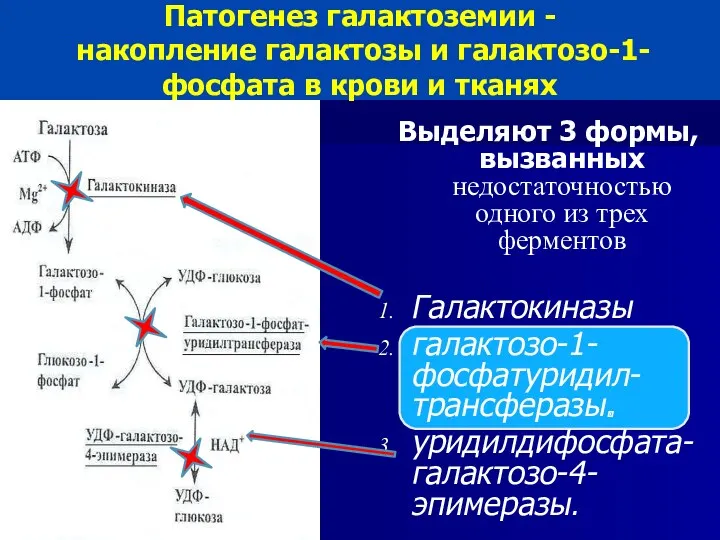
Патогенез галактоземии -
накопление галактозы и галактозо-1-фосфата в крови и тканях
Патогенез галактоземии - накопление галактозы и галактозо-1-фосфата в крови и тканях
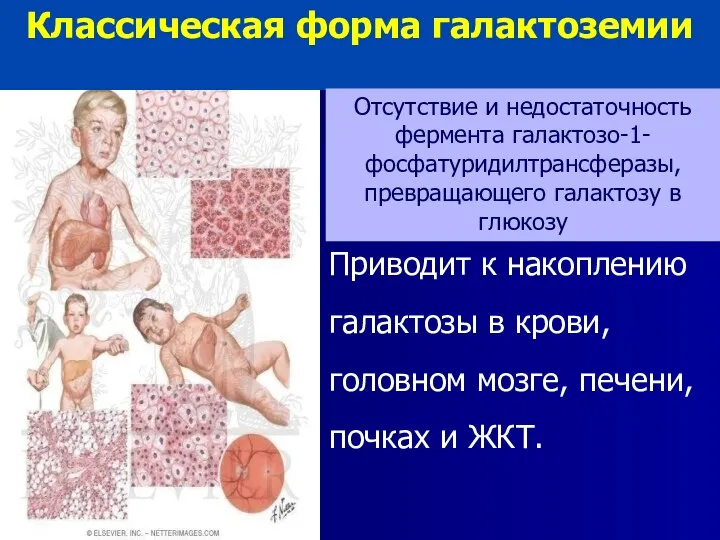
Приводит к накоплению галактозы в крови, головном мозге, печени, почках и
Приводит к накоплению галактозы в крови, головном мозге, печени, почках и

Острое начало на 1-м месяце жизни при кормлении молоком:
Неукротимая рвота, понос,
Острое начало на 1-м месяце жизни при кормлении молоком:
Неукротимая рвота, понос,

Общий анализ крови -анемия.
Биохимия крови:
Повышение уровня: галактозы , АСТ, АЛТ;
Общий анализ крови -анемия.
Биохимия крови:
Повышение уровня: галактозы , АСТ, АЛТ;

Лечение галактоземии
Диета с исключением галактозы.
Замена грудного и коровьего молока, молочных
Лечение галактоземии
Диета с исключением галактозы.
Замена грудного и коровьего молока, молочных
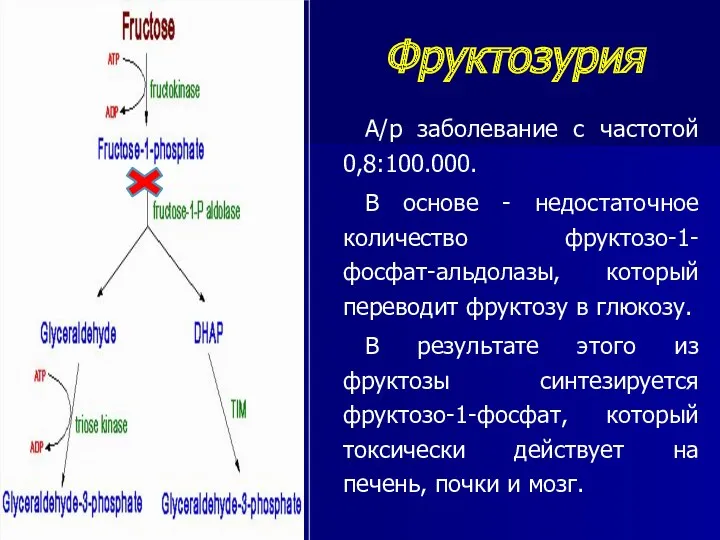
Фруктозурия
А/р заболевание с частотой 0,8:100.000.
В основе - недостаточное количество фруктозо-1-фосфат-альдолазы,
Фруктозурия
А/р заболевание с частотой 0,8:100.000.
В основе - недостаточное количество фруктозо-1-фосфат-альдолазы,

В зависимости от степени недостатка ферментов выделяют легкие и тяжелые формы.
Легкие формы – симптомы незначительные, пациент иногда
В зависимости от степени недостатка ферментов выделяют легкие и тяжелые формы. Легкие формы – симптомы незначительные, пациент иногда

Клиника
Гипогликемические состояния после приема пищи, содержащей фруктозу;
увеличении печени с биохимическими
Клиника
Гипогликемические состояния после приема пищи, содержащей фруктозу;
увеличении печени с биохимическими

Диагностика
Проба с нагрузкой фруктозой вызывает гипогликемическую реакцию. Условием ее проведения должна
Диагностика
Проба с нагрузкой фруктозой вызывает гипогликемическую реакцию. Условием ее проведения должна

Лечение
Диета с исключением из рациона ребенка «вредных» продуктов питания, не менее
Лечение
Диета с исключением из рациона ребенка «вредных» продуктов питания, не менее

Наследственные болезни обмена
ЛИПИДОВ
К этим заболеваниям относятся липидозы и
Наследственные болезни обмена
ЛИПИДОВ
К этим заболеваниям относятся липидозы и
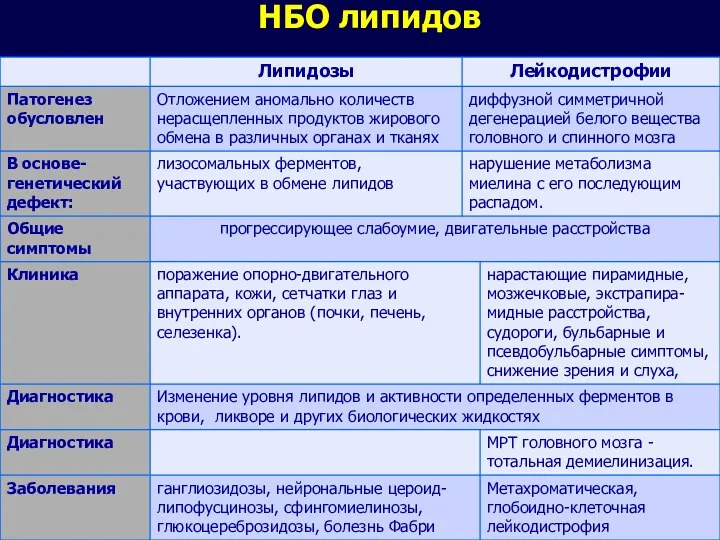
НБО липидов
НБО липидов

Амавротическая идиотия
— редкое наследственное заболевание с АР типом наследования, при
Амавротическая идиотия
— редкое наследственное заболевание с АР типом наследования, при

Частота встречаемости заболевания: 1:300.000 (среди евреев ашкинази– 1:3600).
Около 3% населения
Частота встречаемости заболевания: 1:300.000 (среди евреев ашкинази– 1:3600).
Около 3% населения

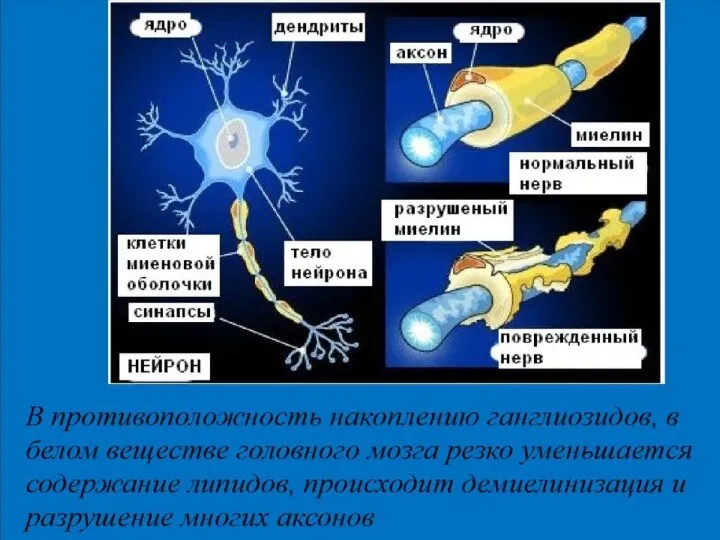
Патогенез болезни
Тея — Сакса
генетический дефект гена, приводит к отсутствию фермента
Патогенез болезни
Тея — Сакса
генетический дефект гена, приводит к отсутствию фермента
Выделяют 4 формы в зависимости от времени начала заболевания и клиники.
Выделяют 4 формы в зависимости от времени начала заболевания и клиники.

Клиника болезни Тея — Сакса
1. Задержка, а затем регресс развития (после
Клиника болезни Тея — Сакса
1. Задержка, а затем регресс развития (после
5. Фармакорезистентные судороги
6. Центральные тетрапарезы.
7. МРТ головного мозга: атрофия нейронов
5. Фармакорезистентные судороги
6. Центральные тетрапарезы.
7. МРТ головного мозга: атрофия нейронов
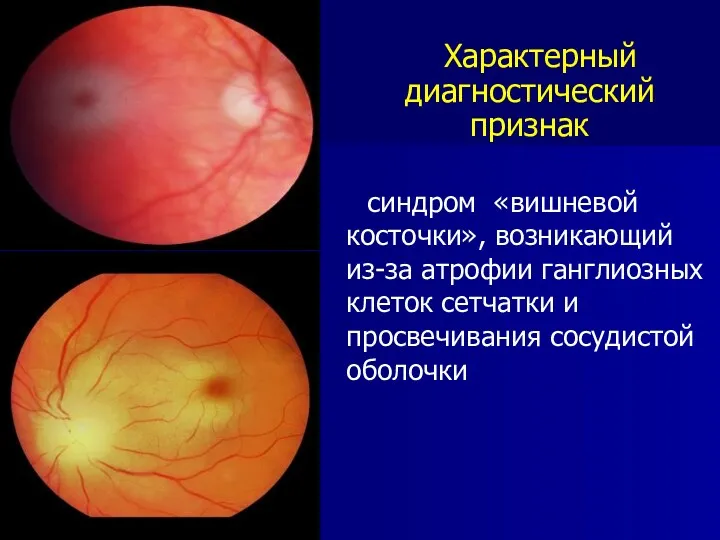
Характерный диагностический признак
синдром «вишневой косточки», возникающий из-за атрофии ганглиозных клеток
синдром «вишневой косточки», возникающий из-за атрофии ганглиозных клеток

Диагностика, лечение, прогноз
В плазме крови:
увеличено содержание цереброзидов и сульфатидов;
В Er
В плазме крови:
увеличено содержание цереброзидов и сульфатидов;
В Er

НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ
лизосомные болезни накопления
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ
лизосомные болезни накопления
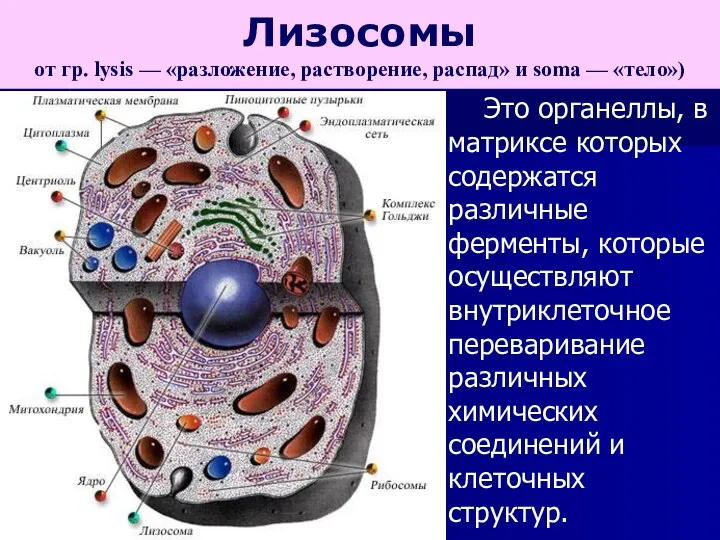
Это органеллы, в матриксе которых содержатся различные ферменты, которые осуществляют внутриклеточное
Это органеллы, в матриксе которых содержатся различные ферменты, которые осуществляют внутриклеточное

Лизосомные болезни накопления (ЛБН)
Обусловлены мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул
Лизосомные болезни накопления (ЛБН)
Обусловлены мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул

Лизосомные болезни накопления(ЛБН)
Число известных форм – около 50
Наследуются по аутосомно-рецессивному типу
Лизосомные болезни накопления(ЛБН)
Число известных форм – около 50
Наследуются по аутосомно-рецессивному типу
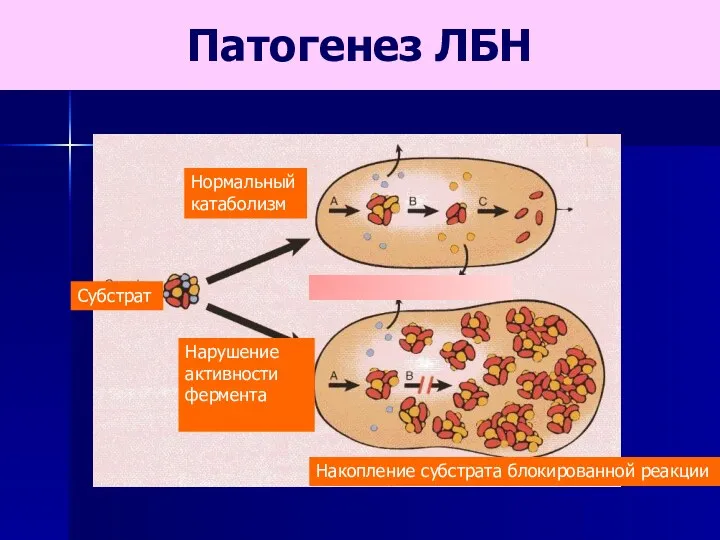
Нормальный
катаболизм
Субстрат
Нарушение активности фермента
Накопление субстрата блокированной реакции
Патогенез ЛБН
Нормальный
катаболизм
Субстрат
Нарушение активности фермента
Накопление субстрата блокированной реакции
Патогенез ЛБН

Классификация ЛБН
В зависимости от накапливаемых субстратов:
Сфинголипидозы (болезнь Гоше, метахроматическая лейкодистрофия, болезнь
Классификация ЛБН
В зависимости от накапливаемых субстратов:
Сфинголипидозы (болезнь Гоше, метахроматическая лейкодистрофия, болезнь
Лизосомальные болезни обмена
Лизосомальные болезни обмена

Клинические проявления ЛБН
Прогрессирующий характер заболевания
Наличие различного по продолжительности интервала нормального развития
Клинические проявления ЛБН
Прогрессирующий характер заболевания
Наличие различного по продолжительности интервала нормального развития

МУКОПОЛИСАХАРИДОЗЫ
МУКОПОЛИСАХАРИДОЗЫ
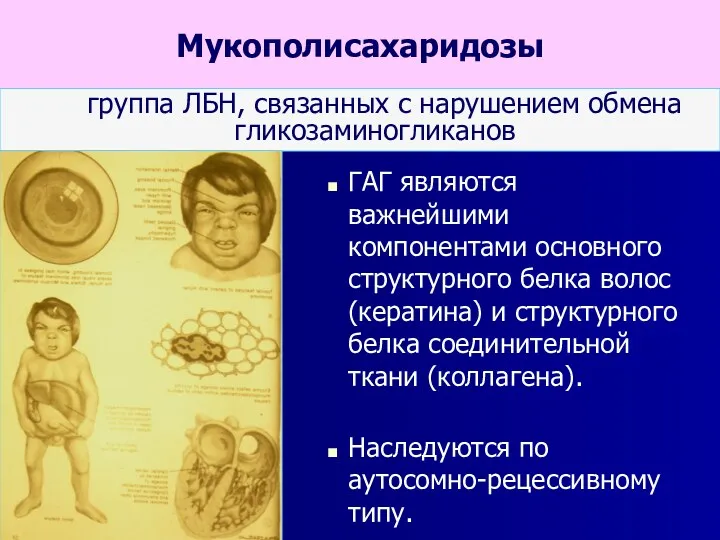
Мукополисахаридозы
группа ЛБН, связанных с нарушением обмена гликозаминогликанов
ГАГ являются важнейшими
Мукополисахаридозы
группа ЛБН, связанных с нарушением обмена гликозаминогликанов
ГАГ являются важнейшими

Патогенез МПС I типа
Недостаточность фермента α-L-идуронидазы
Нарушение отщепления терминального остатка идуроновой кислоты
Патогенез МПС I типа
Недостаточность фермента α-L-идуронидазы
Нарушение отщепления терминального остатка идуроновой кислоты
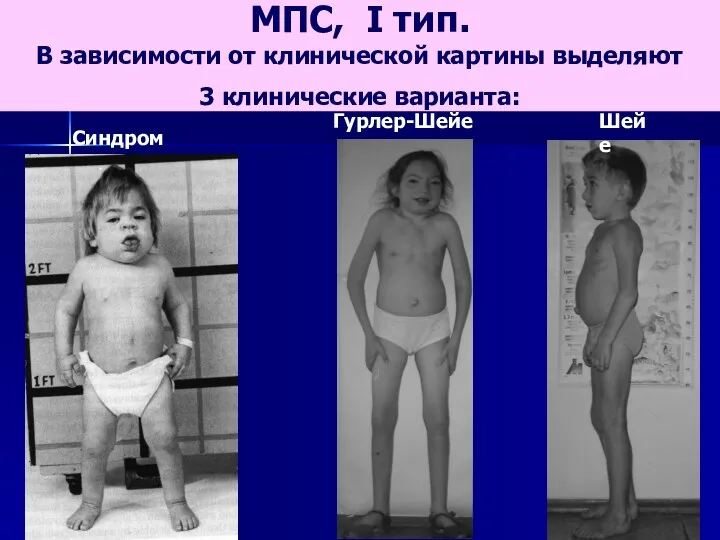
МПС, I тип.
В зависимости от клинической картины выделяют
3 клинические
МПС, I тип. В зависимости от клинической картины выделяют 3 клинические
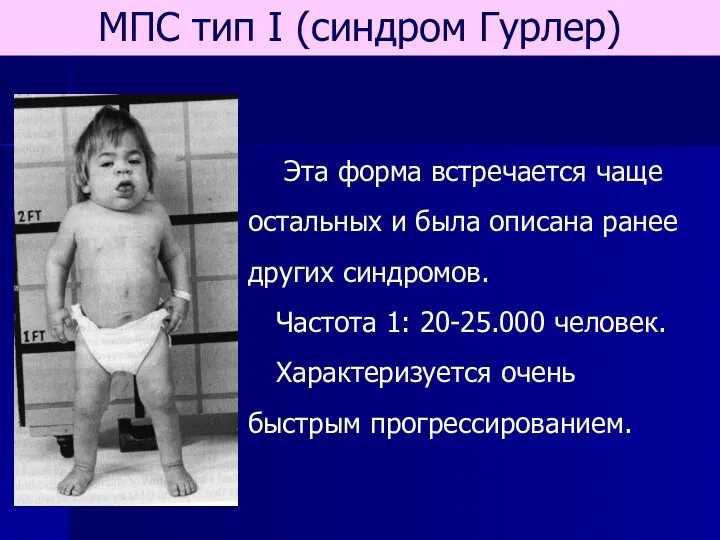
МПС тип I (синдром Гурлер)
Эта форма встречается чаще остальных и
МПС тип I (синдром Гурлер)
Эта форма встречается чаще остальных и
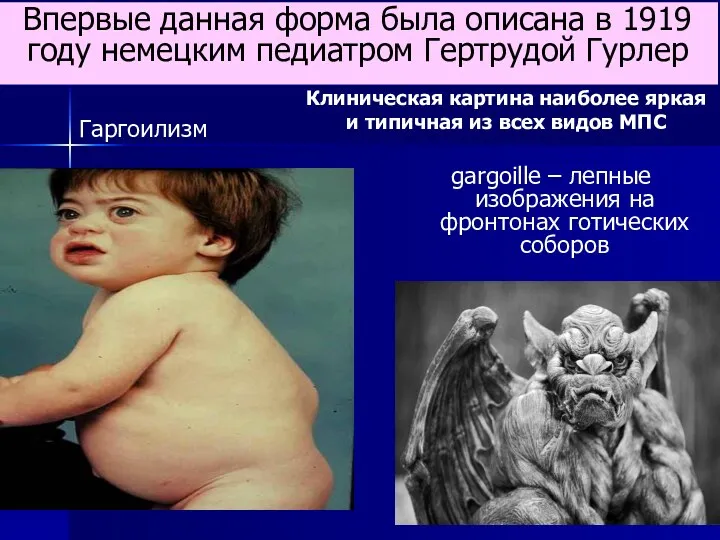
gargoille – лепные изображения на фронтонах готических соборов
Впервые данная форма была
gargoille – лепные изображения на фронтонах готических соборов
Впервые данная форма была
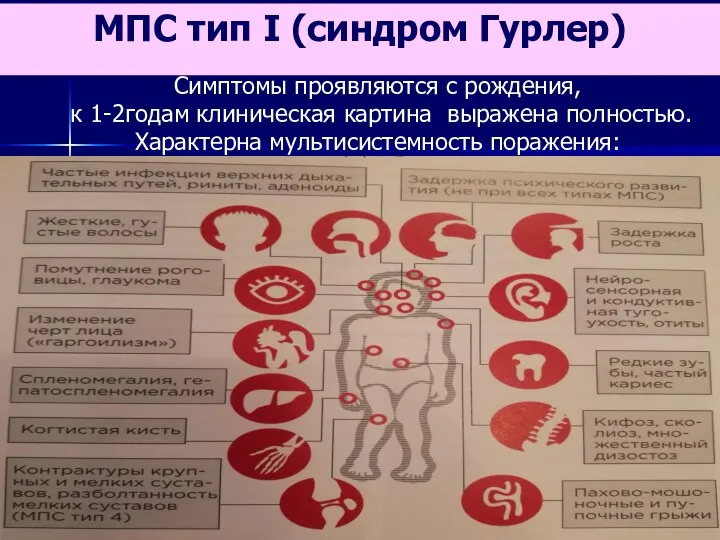
Симптомы проявляются с рождения,
к 1-2годам клиническая картина выражена полностью.
к 1-2годам клиническая картина выражена полностью.
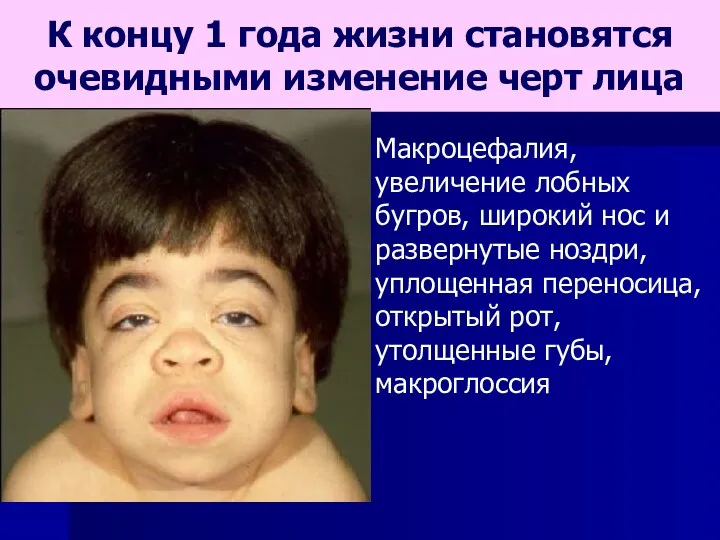
К концу 1 года жизни становятся очевидными изменение черт лица
Макроцефалия,
увеличение
К концу 1 года жизни становятся очевидными изменение черт лица
Макроцефалия,
увеличение
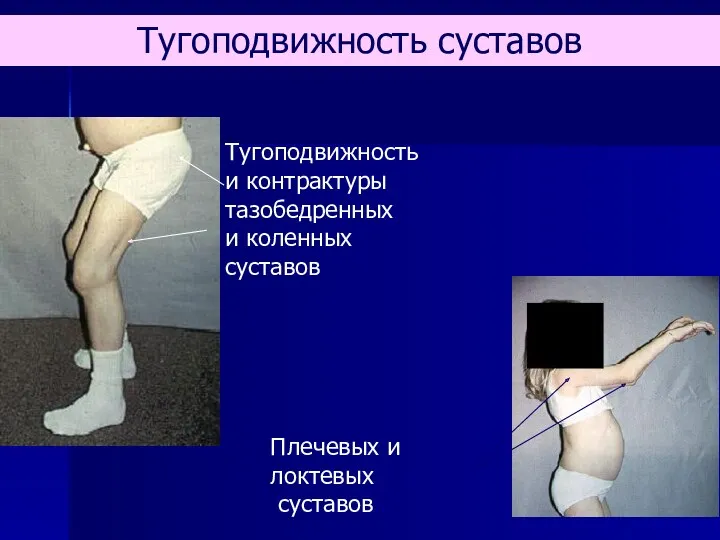
Тугоподвижность
и контрактуры
тазобедренных
и коленных суставов
Плечевых и локтевых
суставов
Тугоподвижность суставов
Тугоподвижность
и контрактуры
тазобедренных
и коленных суставов
Плечевых и локтевых
суставов
Тугоподвижность суставов
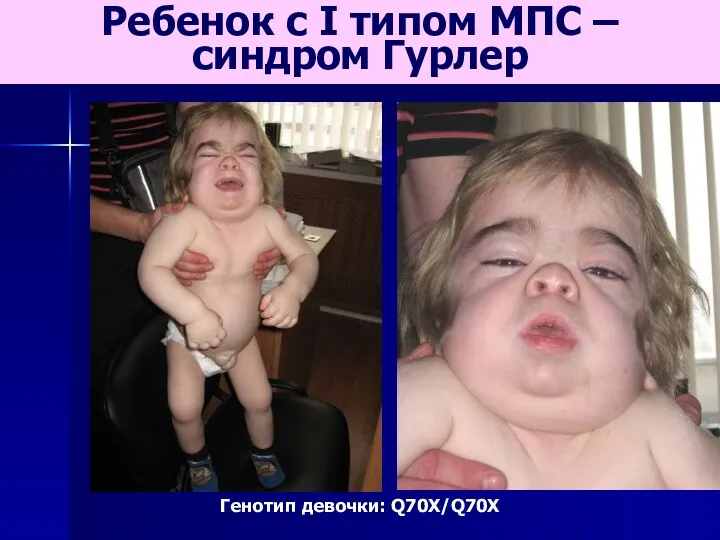
Ребенок с I типом МПС –
синдром Гурлер
Генотип девочки: Q70X/Q70X
Ребенок с I типом МПС –
синдром Гурлер
Генотип девочки: Q70X/Q70X

Больные больше похожи друг на друга, чем на своих здоровых братьев
Больные больше похожи друг на друга, чем на своих здоровых братьев

Синдром Гурлер
манифестирует на 1 году жизни
тяжелая соматическая и неврологическая патология
Синдром Гурлер—Шейе
манифестирует
Синдром Гурлер
манифестирует на 1 году жизни
тяжелая соматическая и неврологическая патология
Синдром Гурлер—Шейе
манифестирует

Дети 3,5 и 5 лет с синдромами Гурлер (слева) и Гурлер-Шейе
Дети 3,5 и 5 лет с синдромами Гурлер (слева) и Гурлер-Шейе

Диагностика МПС
характерные клинические проявления,
определении экскреции с мочой ГАГ
пренатальная диагностика (определение
Диагностика МПС
характерные клинические проявления,
определении экскреции с мочой ГАГ
пренатальная диагностика (определение

Фермент-заместительная терапия :
альдуразим при МПС 1 типа,
элапраза при МПС
Фермент-заместительная терапия :
альдуразим при МПС 1 типа,
элапраза при МПС

Болезнь Ниманна-Пика
Редкое наследственное аутосомно-рецессивное заболевание.
Частота 1:10.000.
Среди евреев-ашкинази 1:100
Мутации гена
Болезнь Ниманна-Пика
Редкое наследственное аутосомно-рецессивное заболевание.
Частота 1:10.000.
Среди евреев-ашкинази 1:100
Мутации гена
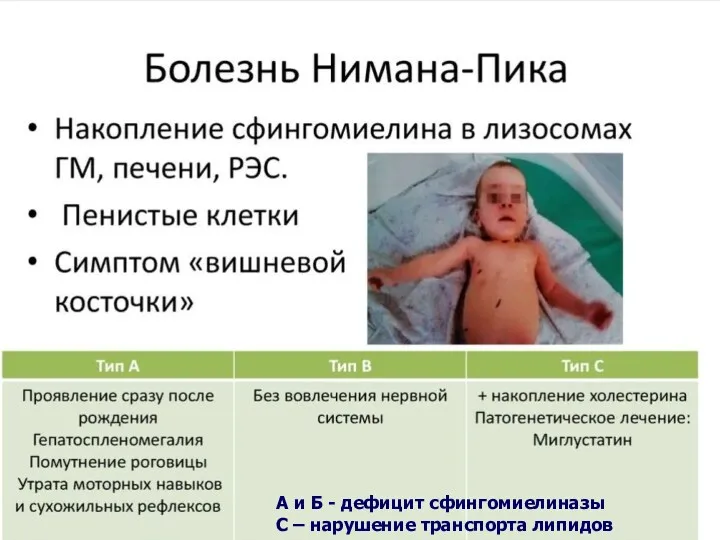
А и Б - дефицит сфингомиелиназы
С – нарушение транспорта липидов
А и Б - дефицит сфингомиелиназы
С – нарушение транспорта липидов

Болезнь Ниманна-Пика А
Ранняя манифестация заболевания
Гепатоспленомегалия
Прогрессирующая церебеллярная атаксия
Снижение интеллекта.
Вертикальный
Болезнь Ниманна-Пика А
Ранняя манифестация заболевания
Гепатоспленомегалия
Прогрессирующая церебеллярная атаксия
Снижение интеллекта.
Вертикальный
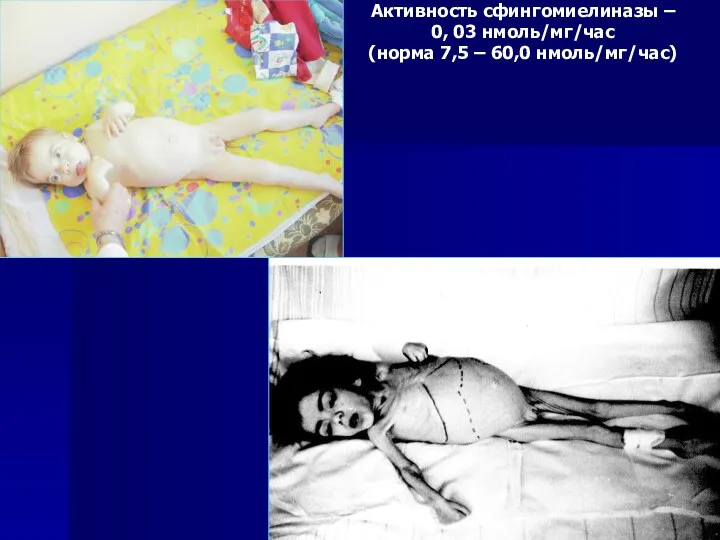
Активность сфингомиелиназы –
0, 03 нмоль/мг/час
(норма 7,5 – 60,0 нмоль/мг/час)
Активность сфингомиелиназы –
0, 03 нмоль/мг/час
(норма 7,5 – 60,0 нмоль/мг/час)

Болезнь Гоше
Мутация в гене GBA приводит к дефициту фермента- глюкоцереброзидазы, в
Болезнь Гоше
Мутация в гене GBA приводит к дефициту фермента- глюкоцереброзидазы, в
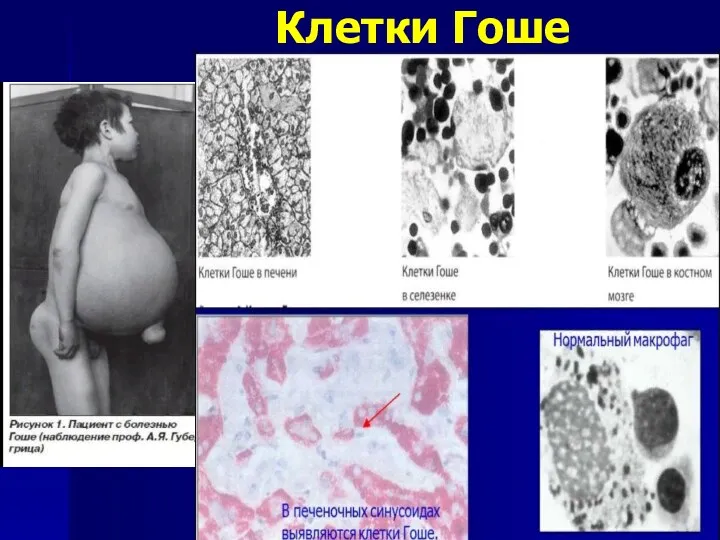
Клетки Гоше
Клетки Гоше
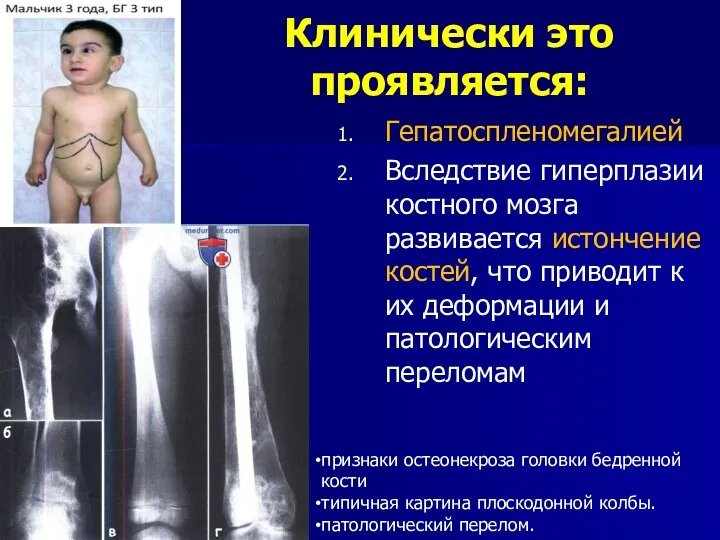
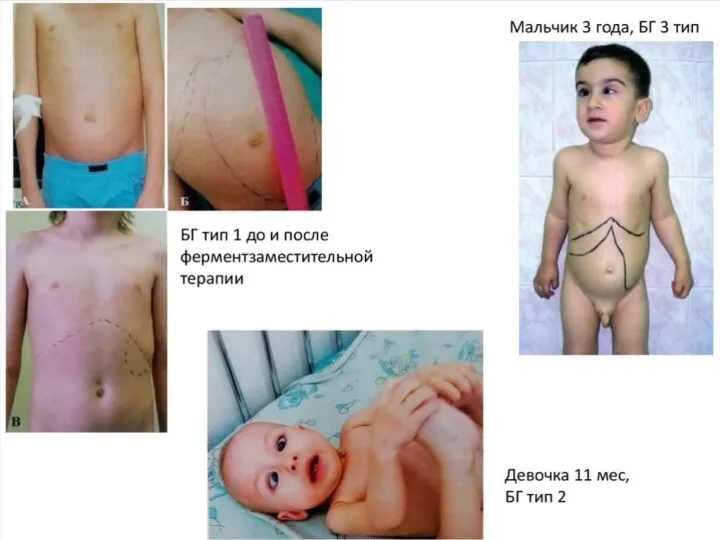
Клинически это проявляется:
Гепатоспленомегалией
Вследствие гиперплазии костного мозга развивается истончение костей, что приводит
Клинически это проявляется:
Гепатоспленомегалией
Вследствие гиперплазии костного мозга развивается истончение костей, что приводит

Клиника
3. Поражение костного мозга сопровождается панцитопенией.
4. Поражение ЦНС сопровождается
судорогами, умственной
Клиника
3. Поражение костного мозга сопровождается панцитопенией.
4. Поражение ЦНС сопровождается
судорогами, умственной
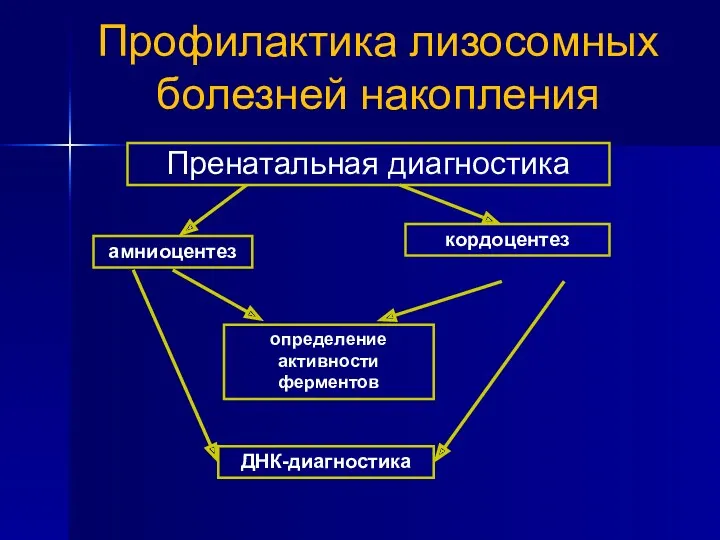
Профилактика лизосомных болезней накопления
Пренатальная диагностика
амниоцентез
кордоцентез
определение активности ферментов
ДНК-диагностика
Профилактика лизосомных болезней накопления
Пренатальная диагностика
амниоцентез
кордоцентез
определение активности ферментов
ДНК-диагностика

Основные термины
медицинской генетики
Основные термины
медицинской генетики

Генетическая гетерогенность -
разнообразие генетических причин
наследственных болезней
- феномен, когда клинически
Генетическая гетерогенность -
разнообразие генетических причин
наследственных болезней
- феномен, когда клинически

Причины генотипического полиморфизма:
различные гены контролируют различные звенья одного и того же
Причины генотипического полиморфизма:
различные гены контролируют различные звенья одного и того же

Генотипический полиморфизм приводит к:
многообразию отдельных симптомов;
изменчивости этих признаков;
появлению переходных
Генотипический полиморфизм приводит к:
многообразию отдельных симптомов;
изменчивости этих признаков;
появлению переходных

Плейотропное действие – множественное действие гена, когда один ген влияет на
Плейотропное действие – множественное действие гена, когда один ген влияет на

Плейотропное действие
Плейотропное проявление мутантного гена, обнаруживается практически при всех наследственных заболеваниях.
Серповидно-клеточная
Плейотропное действие
Плейотропное проявление мутантного гена, обнаруживается практически при всех наследственных заболеваниях.
Серповидно-клеточная

Пенетрантность генов - частота проявления того или иного гена, измеряемая частотой
Пенетрантность генов - частота проявления того или иного гена, измеряемая частотой
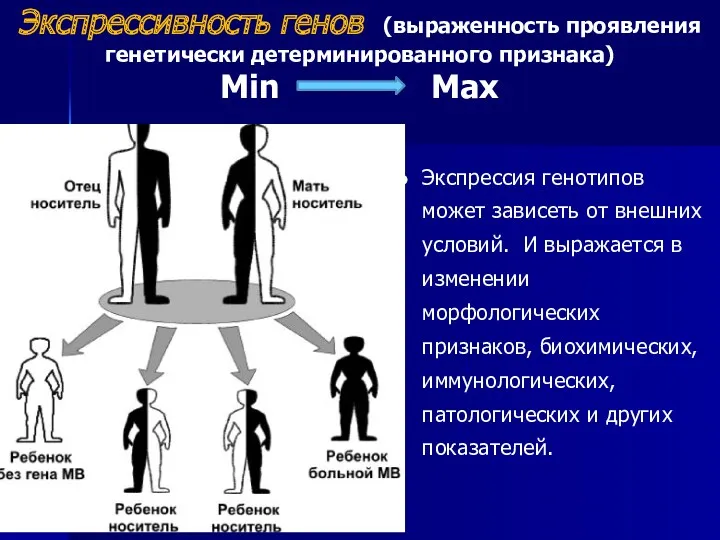
Экспрессивность генов (выраженность проявления генетически детерминированного признака)
Min Max
Экспрессия генотипов может зависеть
Экспрессивность генов (выраженность проявления генетически детерминированного признака)
Min Max
Экспрессия генотипов может зависеть

Экспрессивность генов
Содержание хлора в поте у человека не более 40 ммоль/л,
Экспрессивность генов
Содержание хлора в поте у человека не более 40 ммоль/л,

Гипотеза условного тропизма
дефектных генов (Давыденков С.Н.)
Мутантный патологический ген обнаруживает собственный
Гипотеза условного тропизма
дефектных генов (Давыденков С.Н.)
Мутантный патологический ген обнаруживает собственный

Микропризнаки или
малые аномалии развития
- это стойкий морфологический вариант изменения
Микропризнаки или
малые аномалии развития
- это стойкий морфологический вариант изменения

малые аномалии развития
Не имеют серьезного медицинского или косметического значения, но
малые аномалии развития
Не имеют серьезного медицинского или косметического значения, но

Малые аномалии развития
Диагностическая значимость микропризнаков увеличивается, если они сочетаются друг с
Малые аномалии развития
Диагностическая значимость микропризнаков увеличивается, если они сочетаются друг с
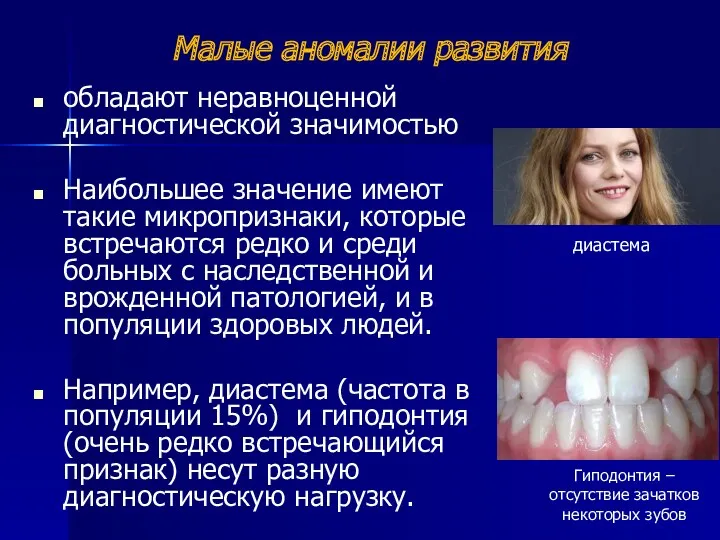
Малые аномалии развития
обладают неравноценной диагностической значимостью
Наибольшее значение имеют такие
Малые аномалии развития
обладают неравноценной диагностической значимостью
Наибольшее значение имеют такие
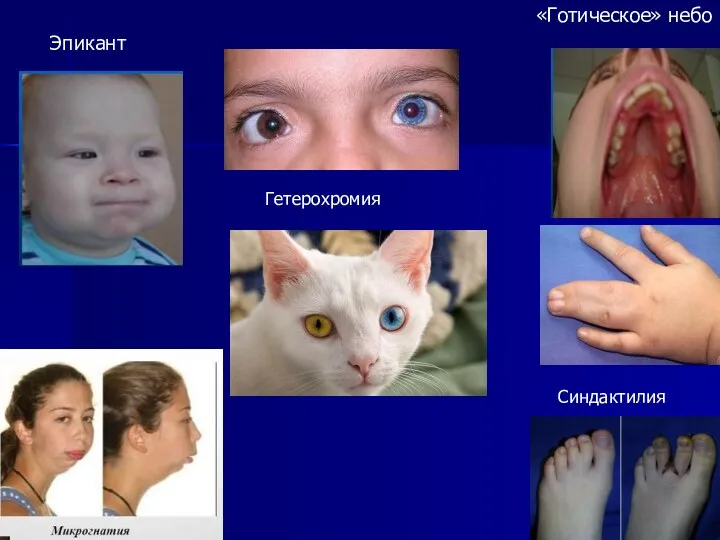
Эпикант
«Готическое» небо
Синдактилия
Гетерохромия
Эпикант
«Готическое» небо
Синдактилия
Гетерохромия

МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
Одна из первых медико-генетических консультаций в мире была создана отечественным
МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
Одна из первых медико-генетических консультаций в мире была создана отечественным
- отрасль профилактической медицины, главной целью которой является снижение количества генетически
- отрасль профилактической медицины, главной целью которой является снижение количества генетически

ЦЕЛЬ ГЕНЕТИЧЕСКОЙ КОНСУЛЬТАЦИИ
установление степени генетического риска;
помощь семье в принятии правильного
ЦЕЛЬ ГЕНЕТИЧЕСКОЙ КОНСУЛЬТАЦИИ
установление степени генетического риска;
помощь семье в принятии правильного
Информация о генетическом риске выдается в виде вероятностей, а степень риска
Информация о генетическом риске выдается в виде вероятностей, а степень риска
Генетический риск определяется двумя способами:
Путем теоретических расчетов, основанных на генетических закономерностях;
С
Генетический риск определяется двумя способами:
Путем теоретических расчетов, основанных на генетических закономерностях;
С

ГЕНЕТИЧЕСКИЙ РИСК:
Низкий (до 5%) – нет противопоказаний к деторождению в семье;
Средний
ГЕНЕТИЧЕСКИЙ РИСК:
Низкий (до 5%) – нет противопоказаний к деторождению в семье;
Средний
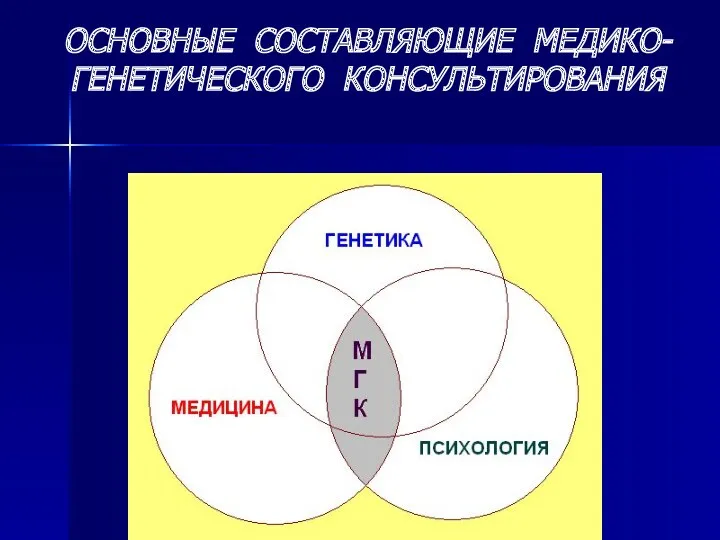
ОСНОВНЫЕ СОСТАВЛЯЮЩИЕ МЕДИКО-ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ
ОСНОВНЫЕ СОСТАВЛЯЮЩИЕ МЕДИКО-ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ

ПОКАЗАНИЯ для медико-генетического консультирования:
Наследственный дефект подозревается, и для уточнения диагноза
ПОКАЗАНИЯ для медико-генетического консультирования:
Наследственный дефект подозревается, и для уточнения диагноза

Отставание ребенка в физическом или
умственном развитии.
Наличие МАР в сочетании
Отставание ребенка в физическом или
умственном развитии.
Наличие МАР в сочетании

Кровное родство родителей больного ребенка.
Повторные случаи мертворождения в семье при отсутствии
Кровное родство родителей больного ребенка.
Повторные случаи мертворождения в семье при отсутствии

Этапы медико-генетического консультирования
Диагноз
Прогноз
Заключение
Совет
ВСЕ РЕШЕНИЯ ПО ПЛАНИРОВАНИЮ СЕМЬИ ПРИНИМАЮТСЯ ТОЛЬКО СУПРУГАМИ
Этапы медико-генетического консультирования
Диагноз
Прогноз
Заключение
Совет
ВСЕ РЕШЕНИЯ ПО ПЛАНИРОВАНИЮ СЕМЬИ ПРИНИМАЮТСЯ ТОЛЬКО СУПРУГАМИ

Первичная профилактика наследственных болезней
1. Планирование деторождения
Оптимальный возраст для женщины 21-35 лет;
Отказ
Первичная профилактика наследственных болезней
1. Планирование деторождения
Оптимальный возраст для женщины 21-35 лет;
Отказ

Опасность близкородственных браков
У них родились 10 детей.
3 из которых умерли
Опасность близкородственных браков
У них родились 10 детей.
3 из которых умерли
Вторичная профилактика
прерывание беременности при высоком риске наследственного заболевания, либо при
Вторичная профилактика
прерывание беременности при высоком риске наследственного заболевания, либо при
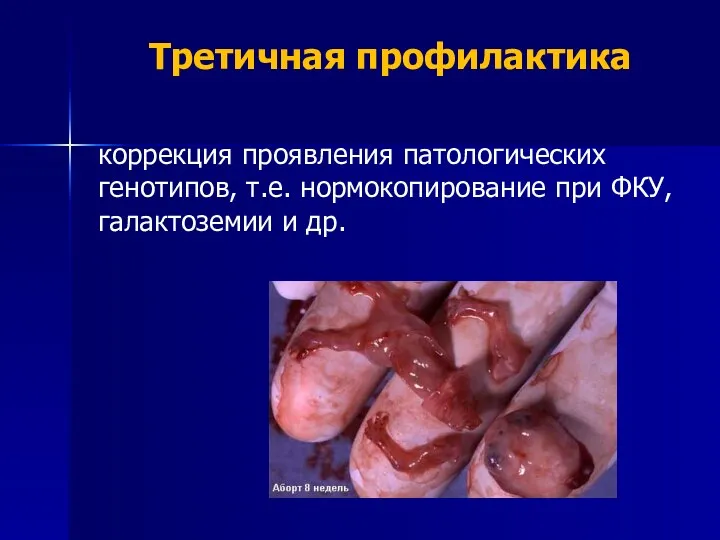
Третичная профилактика
коррекция проявления патологических генотипов, т.е. нормокопирование при ФКУ, галактоземии
Третичная профилактика
коррекция проявления патологических генотипов, т.е. нормокопирование при ФКУ, галактоземии
 Невроз. Виды невроза
Невроз. Виды невроза Аномальные маточные кровотечения: современные подходы лечения и профилактики
Аномальные маточные кровотечения: современные подходы лечения и профилактики ҰлпАның қабынбалы аурулары
ҰлпАның қабынбалы аурулары Қуық және ен қосалқыларының аномалиялары
Қуық және ен қосалқыларының аномалиялары Воспаление висцеральной и париетальной брюшины малого таза - пельвиоперитонит
Воспаление висцеральной и париетальной брюшины малого таза - пельвиоперитонит Антигены и иммунная система человека. Антитела. Строение и функции иммуноглобулинов
Антигены и иммунная система человека. Антитела. Строение и функции иммуноглобулинов Доброкачественные опухоли яичника
Доброкачественные опухоли яичника Аналіз лікувально-діагностичної роботи з хворими на психічні розлади в 7 соматопсихіатричному відділенні
Аналіз лікувально-діагностичної роботи з хворими на психічні розлади в 7 соматопсихіатричному відділенні Доказательная медицина
Доказательная медицина Нашақорлық пен токсикомания эпидемиологиясы. Тәуелділік аурулары кезіндегі соматикалық бұзылыстар
Нашақорлық пен токсикомания эпидемиологиясы. Тәуелділік аурулары кезіндегі соматикалық бұзылыстар Методы исследования физического развития человека
Методы исследования физического развития человека Туберкулез кожи
Туберкулез кожи Общие понятия о пылевых болезнях легких. Пневмокониозы
Общие понятия о пылевых болезнях легких. Пневмокониозы Влияние биоритмов на проявление действия лекарственных средств. Понятие о хронофармакологии
Влияние биоритмов на проявление действия лекарственных средств. Понятие о хронофармакологии Правила забудови, техніка безпеки в операційному блоці
Правила забудови, техніка безпеки в операційному блоці Аномальді бүйректің гистоморфологиялық сипаттамасы
Аномальді бүйректің гистоморфологиялық сипаттамасы Желудочно-кишечные кровотечения
Желудочно-кишечные кровотечения E РОЖИСТОЕ ВОСПАЛЕНИЕ
E РОЖИСТОЕ ВОСПАЛЕНИЕ Общая нозология
Общая нозология Правила наложения повязок на раны
Правила наложения повязок на раны Детский дом-интернат для умственно отсталых детей №15
Детский дом-интернат для умственно отсталых детей №15 Хронические гепатиты у детей
Хронические гепатиты у детей Microlife – уникальные технологии, которые спасают жизни!
Microlife – уникальные технологии, которые спасают жизни! Аллергия – бұл ағзадағы тіндердің өзіндік зақымдалуымен жүретін иммунды реакция болып табылады
Аллергия – бұл ағзадағы тіндердің өзіндік зақымдалуымен жүретін иммунды реакция болып табылады Мұрын және мұрын қойнауларының құрылымы мен топографиясының жасқа байланысты ерекшеліктері және олардың мұрын
Мұрын және мұрын қойнауларының құрылымы мен топографиясының жасқа байланысты ерекшеліктері және олардың мұрын СПИД - чума XXI века. Все что должен знать каждый
СПИД - чума XXI века. Все что должен знать каждый Возбудители зоонозных инфекций
Возбудители зоонозных инфекций Ветеринарное акушерство. Бесплодие. Формы бесплодия
Ветеринарное акушерство. Бесплодие. Формы бесплодия