- Наследственные офтальмопатии

Содержание
- 2. Наследственные офтальмопатии представляют собой самую многочисленную группу моногенных заболеваний, причем в большинстве случаев эти болезни носят
- 3. Патология органа зрения входит в структуру многих хромосомных болезней и моногенных синдромов, наиболее известным из которых
- 4. Другим примером является глазокожнный альбинизм, при котором наряду с депигментацией радужки и сетчатки, у больных наблюдается
- 5. Тирозиназа-негативный глазокожный альбинизм (больные братья и здоровый брат и больная сестра)
- 6. Для глазного альбинизма также как и для глазокожного альбинизма характерна генетическая гетерогенность, но самой распространенной является
- 7. Глазной альбинизм (различные клинические варианты)
- 8. В некоторых случаях специфические аномалии глаз используются в качестве диагностических критериев наследственных синдромов
- 9. Голубые склеры при несовершенном остеогенезе
- 10. Подвывих хрусталика при синдроме Марфана
- 11. Кольцо Кайзера-Флейшера при болезни Вильсона-Коновалова
- 12. Во многих случаях аномалии органа зрения могут быть сопутствующими проявлениями других системных заболеваний
- 13. Кровоизлияния в сетчатку при пернициозной анемии, обусловленной дефицитом витамина B12
- 14. Множественные меланоцитарнные невусы радужки при нейрофиброматозе
- 15. Выворот нижнего века из-за рубцевания с раздражением конъюнктивы и выпадением ресниц при ихтиозе
- 16. В глазах можно найти отражение многих внутренних болезней человека
- 17. Двухцветная радужка при болезни Гиршпрунга
- 18. Конъюнктивит и эписклерит при болезни Крона
- 19. Помутнения в виде звезды под передней капсулой при атопическом дерматите
- 20. Патологические процессы при изолированных наследственных офтальмопатиях могут затрагивать отдельно или в разных комбинациях сетчатку, радужку, роговицу,
- 21. Многие формы наследственных ретинопатий ассоциированы с дефектами фоторецепторов, и для них характерна огромная генетическая гетерогенность
- 22. Наиболее тяжелой формой наследственной ретинопатии является врожденный амавроз Лебера при котором наблюдается одновременное поражение фоторецепторов колбочек
- 23. Врожденный амавроз Лебера — это гетерогенная группа аутосомно-рецессивных заболеваний, характеризующихся очень ранним началом дистрофии сетчатки и
- 24. В настоящее время идентифицированы гены для 15 генетических форм заболевания. Наиболее распространенной является форма 10, объясняющая
- 25. Менее злокачественно протекают наследственная дистрофия колбочек и палочек, пигментный ретинит, макулярная дегенерация сетчатки, ночная и цветовая
- 26. Для каждой из этих групп заболеваний характерно перекрывание спектров клинических проявлений. При этом во многих случаях
- 27. Дистрофия колбочек и палочек — это высоко гетерогенная группа наследственных болезней глаз, характеризующихся ранним ухудшением зрения,
- 28. Болезнь сопровождается распространяющейся пигментацией сетчатки и может быть заподозрена при наличии у пациента фотобоязни, нистагма, снижения
- 29. Пигментный ретинит — это еще более гетерогенная группа заболеваний, основными проявлениями которой являются ночная слепота, развитие
- 30. На глазном дне выявляются темные пигментные области ('bone spicules'), кистоидная макулярная эдема, пигментирование стекловидных клеток и
- 31. Пигментный ретинит, или пигментная дегенерация сетчатки
- 32. В 50-60% случаев болезнь наследуется по аутосомно-рецессивному, в 30-40% — по аутосомно-доминантному и в 5-15% —
- 33. В настоящее время идентифицированы гены при 35 рецессивных, немногим более 20 доминантных и двух Х-сцепленных формах
- 34. Х-сцепленные формы являются наиболее частыми и объясняет около 11% всех форм пигментного ретинита. Они обусловлены мутациями
- 35. Частой также является аутосомно-доминантная форма 20, которая объясняет около 5-10% всех случаев пигментного ретинита в Европе
- 36. Макулярная дегенерация сетчатки — это гетерогенная группа моногенных и многофакторных заболеваний, обусловленных прогрессирующей дегенерацией фоторецепторов и
- 37. Макулярная дегенерация сетчатки
- 38. Наиболее частой наследственной формой макулярной дегенерации сетчатки у детей является болезнь Старгардта, дебютирующая в возрасте 7-12
- 39. Болезнь Старгардта (по глазному дну разбросано множество желтоватых пятнышек неправильной формы)
- 40. Широко распространенная возрастная макулопатия, являющаяся основной причиной потери зрения в пожилом и старческом возрасте, в большинстве
- 41. Возрастная макулопатия
- 42. Врожденная стационарная ночная слепота — это клинически полиморфная и генетически гетерогенная группа непрогрессирующих болезней сетчатки, характеризующихся
- 43. В настоящее время идентифицированы гены для 4 аутосомно-рецессивных, 3 аутосомно-доминантных и 3 Х-сцепленных формах врожденной стационарной
- 44. Цветовая слепота — это гетерогенная группа моногенных заболеваний, при которых у больных нарушено цветовое восприятие, причем
- 45. Тотальная цветовая слепота (палочковая монохромазия или полная ахроматопсия) — это гетерогенная группа, состоящая из 4 редких
- 46. Голубой пигмент кодируется геном OPN1SW. Гетерозиготные мутации в этом гене идентифицированы у больных с тритановой цветовой
- 47. Опсины, чувствительные к красному и зеленому цвету, кодируются семейством родственных генов, расположенных в Xq28. Для красного
- 48. Редкая Х-сцепленная рецессивная голубая монохромия или неполная ахроматопсия может быть обусловлена структурными реорганизациями в области локализации
- 49. Дихромное цветовое восприятие относится к тяжелым дефектам зрения, при которых используются только два типа фоторецепторов —
- 50. Аномальная трихромасия, или монохромазия голубых колбочек, протекающая в общем случае гораздо мягче, — это трихромное цветовое
- 51. В каждом из этих двух случаев болезнь обусловлена соответственно мутациями в генах OPN1LW или OPN1MW .
- 52. Итак, успешная идентификация целого ряда генов, ответственных за наследственные формы ретинопатий, была осуществлена потому, что их
- 53. Основным визуальным пигментом палочек является родопсин. Абсорбция фотона родопсином приводит к триггерному переключению сигнальной передачи в
- 54. В фоторецепторах активируемый светом фотопигмент взаимодействует с трансдуцином — G-белком, стимулирующим ГДФ/ГТФ-обмен. Это взаимодействие, в свою
- 55. Важнейшим звеном фототрансдукции является ретиноидный цикл зрения. Опсины ковалентно связаны с маленькими коньюгированными хромофорами — 11-цис-ретинальдегидом,
- 56. Очищение фоторецепторов от транс-ретиноля происходит с помощью специфического АТФ-связывающего трансмембранного транспортера, кодируемого геном ABCA4. Гомозиготные или
- 57. В нейрональной сети сетчатки свет гиперполяризует фоторецепторы и уменьшает выброс глютаматного нейротрансмиттера. При этом сигналы зрения
- 58. Важнейшую роль в фототрансдукции играет синаптическая передача светового сигнала, в реализации которой принимают участие глютаматные рецепторы
- 59. Но не только нарушение фототрансдукции приводит к развитию наследственных ретинопатий
- 60. Наиболее частые формы врожденного амавроза Лебера и Х-сцепленного пигментного ретинита обусловлены цилиарной дисфункцией. К этой же
- 61. К различным вариантам дистрофии колбочек и палочек, пигментного ретинита и макулярной дегенерации сетчатки могут приводить также
- 62. Атрофия зрительного нерва Лебера является классическим примером митохондриальных заболеваний. Клинически болезнь характеризуется острой потерей центрального зрения
- 63. Наследственная нейропатия зрительного нерва Лебера
- 64. Патологические изменения основных структур глазного яблока в большей степени ассоциированы с дефектами белков внеклеточного матрикса или
- 65. Мутации в генах коллагенов, протеогликанов, микрофибриллярных белков или гликопротеинов внеклеточного матрикса лежат в основе развития наследственных
- 66. Мажорный хрящевой коллаген II типа составляет основу стекловидного тела, поэтому его дефекты приводят к дегенеративным процессам
- 67. Её основными клиническими проявлениями является дегенерация стекловидного тела и самопроизвольная отслойка сетчатки. Диагноз болезни Вагнера обычно
- 68. Наследственные дефекты в коллагене VIII типа, участвующем в образовании десцеметовой мембраны, идентифицированы у больных c двумя
- 69. Эпителиально-эндотелиальная дистрофия роговицы Фукса (буллезная кератопатия, отек стромы)
- 70. Задняя полиморфная дистрофия роговицы (утолщение Десцеметовой мембраны)
- 71. Редкий аутосомно-рецессивный синдром Кноблоха (витреоретинальная дегенерация, сочетающаяся с отслоением сетчатки, макулярными аномалиями, тяжелой миопией и затылочным
- 72. Аутосомно-доминантная витреоретинальная дегенерация с поздним началом обусловлена мутациями в гене C1QTNF5, кодирующем белок с коллагено-подобным доменом.
- 73. Целый ряд изолированных офтальмопатий обусловлен наследственными дефектами протеогликанов, участвующих в обеспечении прозрачности роговицы глаза и функционировании
- 74. Ранее мы уже обсуждали роль никталопина в развитии одной из генетических форм врожденной стационарной ночной слепоты.
- 75. Плоская роговица глаза (роговица уплощена,тонкая, передняя камера мелкая, исследование при помощи камеры Шеймпфлюга)
- 76. Кератокан и другие малые кератансульфат-протеогликаны, в первую очередь, люмикан участвуют в регуляции пространственной организации коллагеновых фибрилл.
- 77. Одной из важнейших причин развития наследственных офтальмопатий являются нарушения морфогенеза органа зрения, в контроле которого принимают
- 78. Характерным свойством этой группы заболеваний является огромный клинический полиморфизм, то есть существование длинных аллельных серий, образованных
- 79. Так, не менее 6 аллельных вариантов аутосомно-доминантной дистрофии роговицы обусловлены мутациями в гене кератоэпителина – небольшого
- 80. Это медленно прогрессирующие гранулярные дистрофии I (Гренува) и II (Авеллино) типов, дебютирующие соответственно во второй и
- 81. Гранулярная дистрофия роговицы, Groenouw I (помутнения роговицы в передних слоях стромы в виде «хлопьев снега»)
- 82. Гранулярная дистрофия роговицы II типа Avellino (амилоидные отложения в передних отделах стромы)
- 83. Дистрофия роговицы Reis-Bückler (помутнения, расположенные субэпителиально и на боуменовой мембране
- 84. Сотовидная дистрофия роговицы Thiel-Behnke (помутнения в боуменовой мембране и базальной мембране эпителия создающие сотоподобный рисунок)
- 85. Различные варианты решетчатой дегенерации
- 86. Однако наибольшее количество изолированных наследственных офтальмопатий связаны с дефектами транскрипционной системы контроля морфогенеза органа зрения
- 87. Два генетических варианта полиморфной задней дистрофии роговицы глаза 3 и 1 типов обусловлены мутациями в генах
- 88. Кератоконус
- 89. Не менее 10 аллельных вариантов аутосомно-доминантных болезней глаз связаны с мутациями в гене транскрипционного фактора Pax6,
- 90. Это частичная или полная аниридия, аномалии Петерса (недоразвитие стромы роговицы, синехии между радужкой и роговицей и
- 91. Аниридия — полное отсутствие радужки
- 92. Аниридия со вторичной врожденной глаукомой
- 93. Аномалия Петерса (врожденные двухсторонние адгезии между центральными отделами роговицы и хрусталиком, с помутнением роговицы. Развивается глаукома)
- 94. Аномалия диска
- 95. Колобома диска зрительного нерва (вместо диска в данном случае видна глубокая ямка, нервная ткань обнаруживается только
- 96. Поверхностный точечный билатеральный кератит
- 97. Врожденная зонулярная катаракта
- 98. Дефекты развития радужки глаза в сочетании с дегенеративными процессами в роговице, первичной глаукомой и другими аномалиями
- 99. Мутации в гене PITX2 идентифицированы при синдроме Ригера 1 и Аксенфельда-Ригера, иридогониодисгенезе 2, кольцевом дермоиде роговицы
- 100. Аномалия Rieger (от стромы радужки идут мостики к роговице, которые тянутся к зоне перед кольцом Швальбе)
- 101. Синдром Axenfeld-Rieger (атрофия стромы радужки, отсутствие "воротника")
- 102. Мезодермальная дисгенезия роговицы и радужки
- 103. Мутации в гене FOXC2 обнаружены при двух аллельных вариантах лимфедемы, сочетающейся с двойным рядом ресниц или
- 104. Лимфедема век (мешки под глазами возникают вследствие лимфостаза и отека в области нижних век)
- 105. Мутации в гене транскрипционного фактора Sox2, избирательно экспрессирующегося в нервной системе, идентифицированы у больных тяжелой билатеральной
- 107. Скачать презентацию

Наследственные офтальмопатии представляют собой самую многочисленную группу моногенных заболеваний, причем в
Наследственные офтальмопатии представляют собой самую многочисленную группу моногенных заболеваний, причем в

Патология органа зрения входит в структуру многих хромосомных болезней и моногенных
Патология органа зрения входит в структуру многих хромосомных болезней и моногенных

Другим примером является глазокожнный альбинизм,
при котором наряду с депигментацией радужки
Другим примером является глазокожнный альбинизм, при котором наряду с депигментацией радужки

Тирозиназа-негативный глазокожный альбинизм (больные братья и здоровый брат и больная сестра)
Тирозиназа-негативный глазокожный альбинизм (больные братья и здоровый брат и больная сестра)

Для глазного альбинизма
также как и для глазокожного альбинизма характерна
Для глазного альбинизма также как и для глазокожного альбинизма характерна
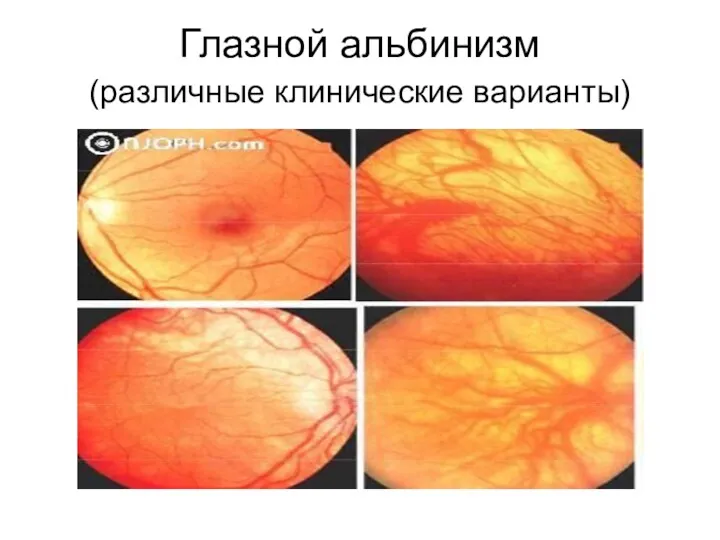
Глазной альбинизм
(различные клинические варианты)
Глазной альбинизм
(различные клинические варианты)

В некоторых случаях специфические аномалии глаз используются в качестве диагностических критериев
В некоторых случаях специфические аномалии глаз используются в качестве диагностических критериев

Голубые склеры при несовершенном остеогенезе
Голубые склеры при несовершенном остеогенезе
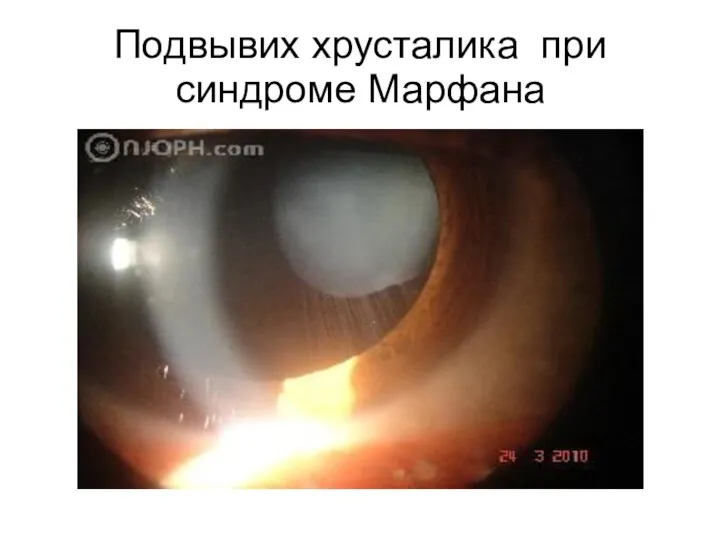
Подвывих хрусталика при синдроме Марфана
Подвывих хрусталика при синдроме Марфана
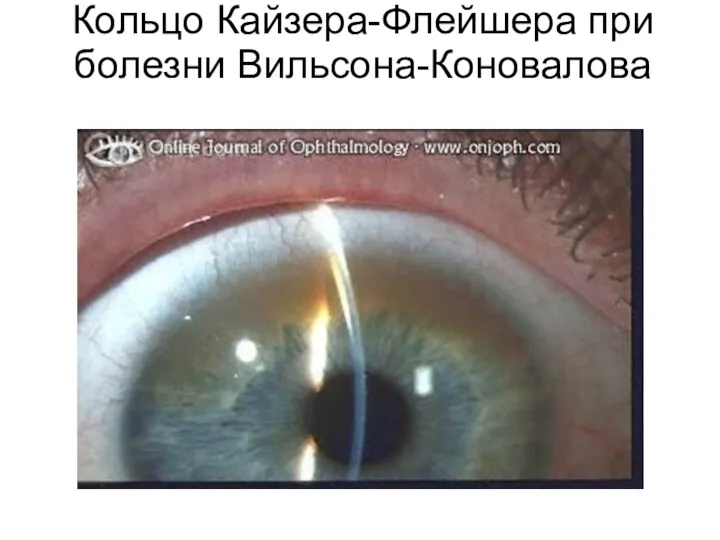
Кольцо Кайзера-Флейшера при болезни Вильсона-Коновалова
Кольцо Кайзера-Флейшера при болезни Вильсона-Коновалова

Во многих случаях аномалии органа зрения могут быть сопутствующими проявлениями других
Во многих случаях аномалии органа зрения могут быть сопутствующими проявлениями других
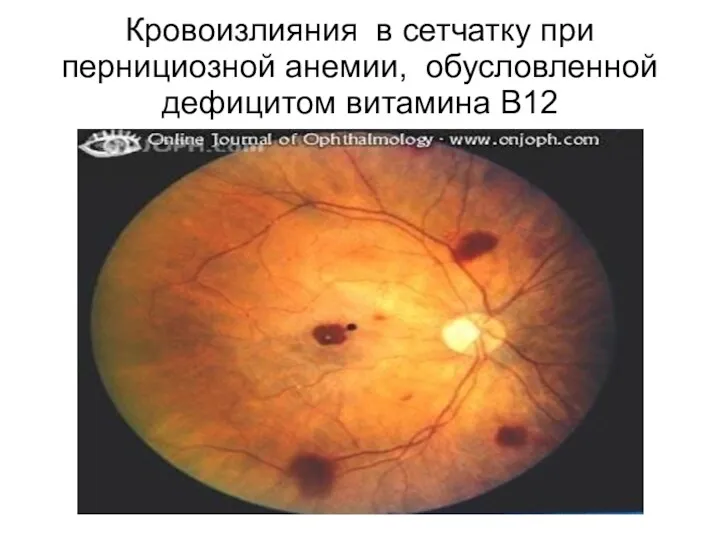
Кровоизлияния в сетчатку при пернициозной анемии, обусловленной дефицитом витамина B12
Кровоизлияния в сетчатку при пернициозной анемии, обусловленной дефицитом витамина B12
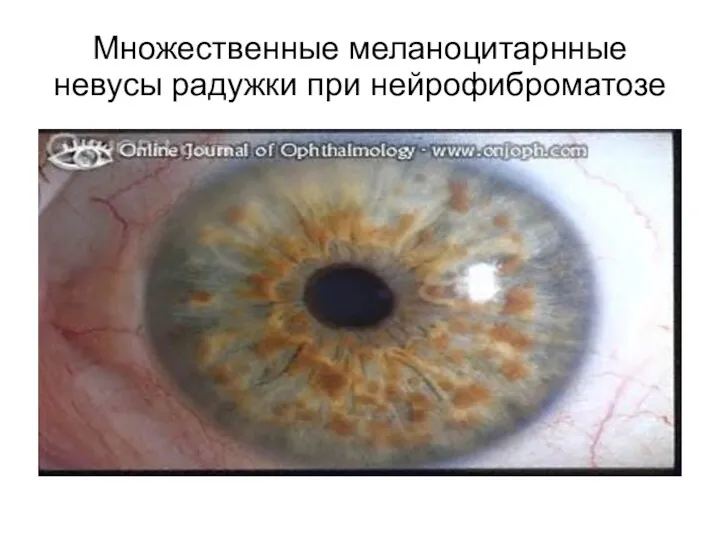
Множественные меланоцитарнные невусы радужки при нейрофиброматозе
Множественные меланоцитарнные невусы радужки при нейрофиброматозе
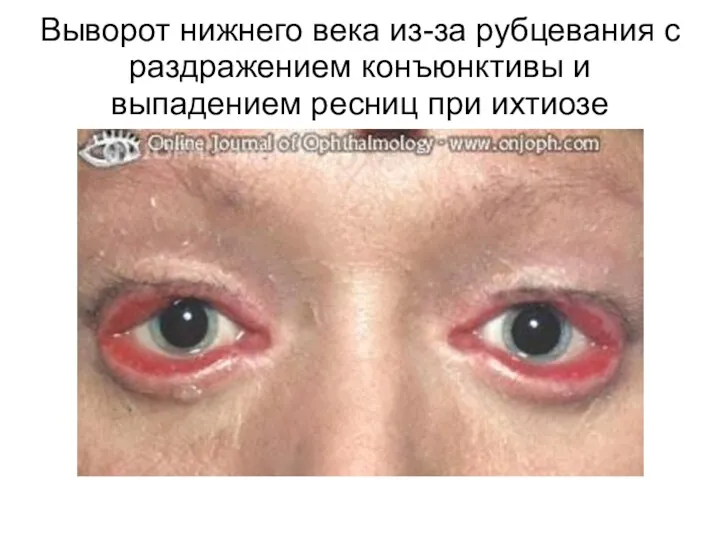
Выворот нижнего века из-за рубцевания с раздражением конъюнктивы и выпадением ресниц
Выворот нижнего века из-за рубцевания с раздражением конъюнктивы и выпадением ресниц

В глазах можно найти отражение многих внутренних болезней человека
В глазах можно найти отражение многих внутренних болезней человека

Двухцветная радужка при болезни Гиршпрунга
Двухцветная радужка при болезни Гиршпрунга
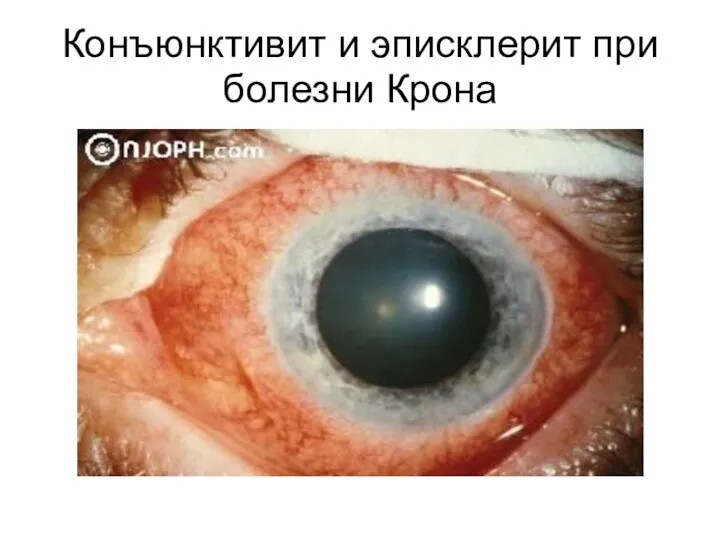
Конъюнктивит и эписклерит при болезни Крона
Конъюнктивит и эписклерит при болезни Крона
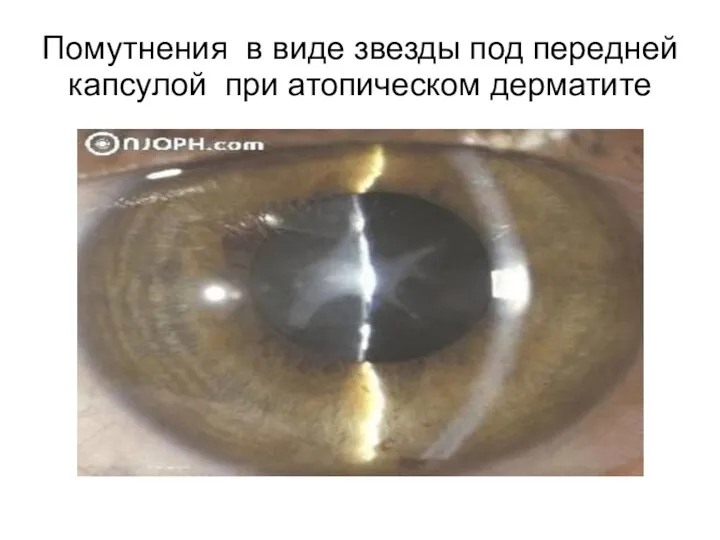
Помутнения в виде звезды под передней капсулой при атопическом дерматите
Помутнения в виде звезды под передней капсулой при атопическом дерматите

Патологические процессы при изолированных наследственных офтальмопатиях могут затрагивать отдельно или в
Патологические процессы при изолированных наследственных офтальмопатиях могут затрагивать отдельно или в

Многие формы наследственных ретинопатий ассоциированы с дефектами фоторецепторов, и для них
Многие формы наследственных ретинопатий ассоциированы с дефектами фоторецепторов, и для них

Наиболее тяжелой формой наследственной ретинопатии является врожденный амавроз Лебера
при котором наблюдается
Наиболее тяжелой формой наследственной ретинопатии является врожденный амавроз Лебера при котором наблюдается

Врожденный амавроз Лебера —
это гетерогенная группа аутосомно-рецессивных заболеваний, характеризующихся очень
Врожденный амавроз Лебера — это гетерогенная группа аутосомно-рецессивных заболеваний, характеризующихся очень

В настоящее время идентифицированы гены для 15 генетических форм заболевания.
Наиболее
В настоящее время идентифицированы гены для 15 генетических форм заболевания. Наиболее

Менее злокачественно протекают наследственная дистрофия колбочек и палочек, пигментный ретинит, макулярная
Менее злокачественно протекают наследственная дистрофия колбочек и палочек, пигментный ретинит, макулярная

Для каждой из этих групп заболеваний характерно перекрывание спектров клинических
Для каждой из этих групп заболеваний характерно перекрывание спектров клинических

Дистрофия колбочек и палочек — это высоко гетерогенная группа наследственных болезней
Дистрофия колбочек и палочек — это высоко гетерогенная группа наследственных болезней

Болезнь сопровождается распространяющейся пигментацией сетчатки и может быть заподозрена при наличии
Болезнь сопровождается распространяющейся пигментацией сетчатки и может быть заподозрена при наличии

Пигментный ретинит — это еще более гетерогенная группа заболеваний, основными проявлениями
Пигментный ретинит — это еще более гетерогенная группа заболеваний, основными проявлениями

На глазном дне выявляются темные пигментные области ('bone spicules'), кистоидная
На глазном дне выявляются темные пигментные области ('bone spicules'), кистоидная
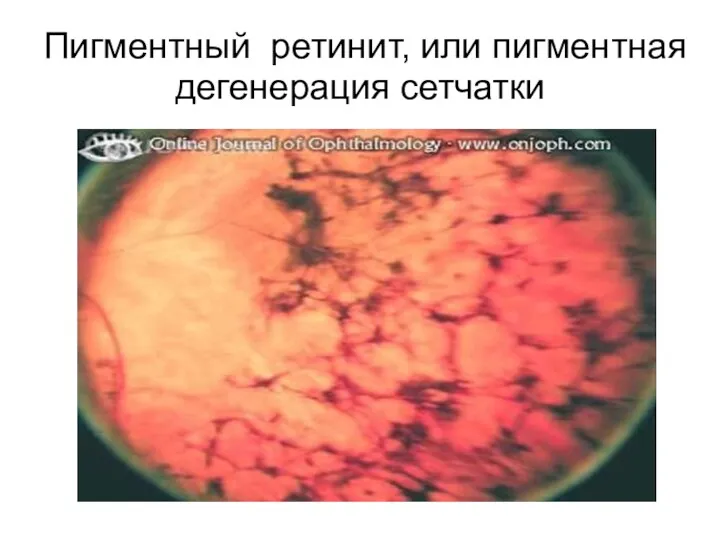
Пигментный ретинит, или пигментная дегенерация сетчатки
Пигментный ретинит, или пигментная дегенерация сетчатки

В 50-60% случаев болезнь наследуется по аутосомно-рецессивному,
в 30-40% — по
В 50-60% случаев болезнь наследуется по аутосомно-рецессивному, в 30-40% — по

В настоящее время идентифицированы гены при 35 рецессивных, немногим более 20
В настоящее время идентифицированы гены при 35 рецессивных, немногим более 20

Х-сцепленные формы являются наиболее частыми и объясняет около 11% всех форм
Х-сцепленные формы являются наиболее частыми и объясняет около 11% всех форм

Частой также является аутосомно-доминантная форма 20, которая объясняет около 5-10% всех
Частой также является аутосомно-доминантная форма 20, которая объясняет около 5-10% всех

Макулярная дегенерация сетчатки — это гетерогенная группа моногенных и многофакторных заболеваний,
Макулярная дегенерация сетчатки — это гетерогенная группа моногенных и многофакторных заболеваний,
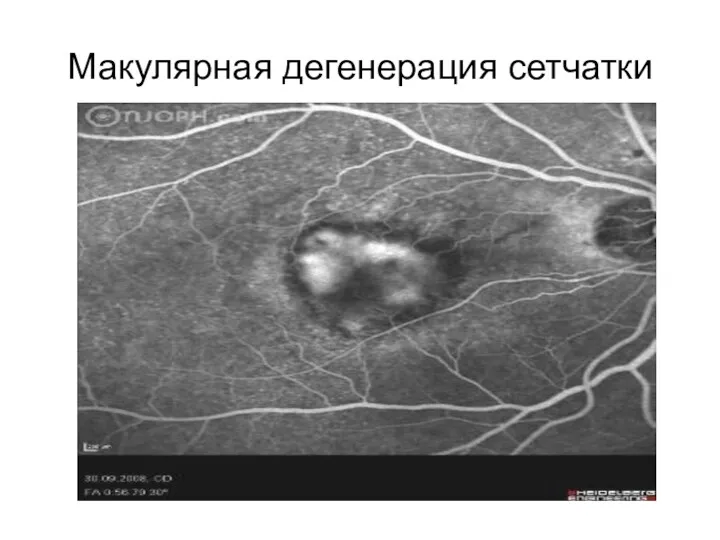
Макулярная дегенерация сетчатки
Макулярная дегенерация сетчатки

Наиболее частой наследственной формой макулярной дегенерации сетчатки у детей является болезнь
Наиболее частой наследственной формой макулярной дегенерации сетчатки у детей является болезнь
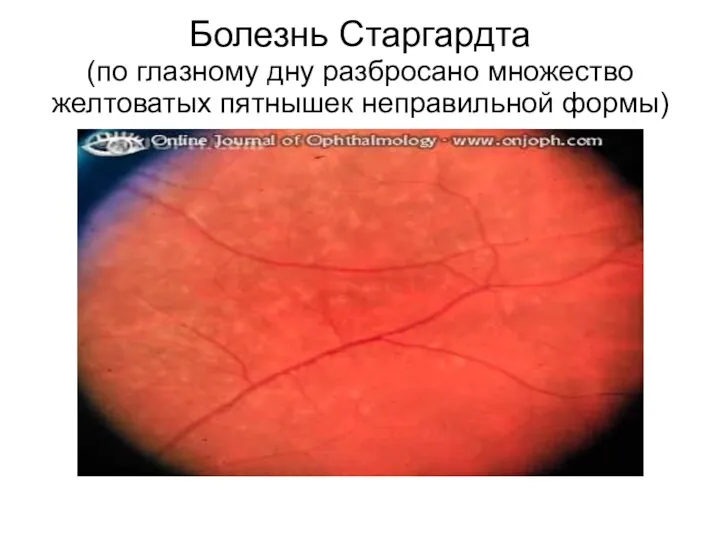
Болезнь Старгардта
(по глазному дну разбросано множество желтоватых пятнышек неправильной формы)
Болезнь Старгардта
(по глазному дну разбросано множество желтоватых пятнышек неправильной формы)

Широко распространенная
возрастная макулопатия,
являющаяся основной причиной потери зрения в пожилом
Широко распространенная возрастная макулопатия, являющаяся основной причиной потери зрения в пожилом
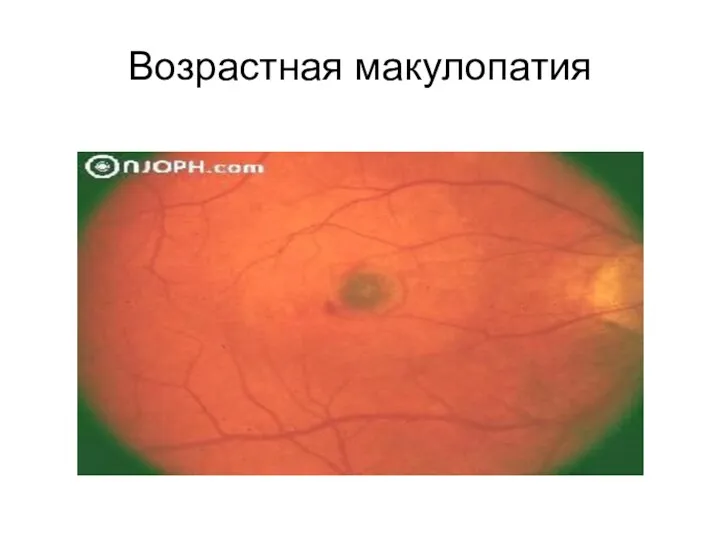
Возрастная макулопатия
Возрастная макулопатия

Врожденная стационарная ночная слепота —
это клинически полиморфная и генетически гетерогенная
Врожденная стационарная ночная слепота — это клинически полиморфная и генетически гетерогенная

В настоящее время идентифицированы гены для 4 аутосомно-рецессивных,
3 аутосомно-доминантных и
В настоящее время идентифицированы гены для 4 аутосомно-рецессивных, 3 аутосомно-доминантных и

Цветовая слепота —
это гетерогенная группа моногенных заболеваний, при которых у
Цветовая слепота — это гетерогенная группа моногенных заболеваний, при которых у

Тотальная цветовая слепота
(палочковая монохромазия или полная ахроматопсия) — это гетерогенная
Тотальная цветовая слепота (палочковая монохромазия или полная ахроматопсия) — это гетерогенная

Голубой пигмент кодируется геном OPN1SW.
Гетерозиготные мутации в этом гене идентифицированы
Голубой пигмент кодируется геном OPN1SW. Гетерозиготные мутации в этом гене идентифицированы

Опсины, чувствительные к красному и зеленому цвету, кодируются семейством родственных генов,
Опсины, чувствительные к красному и зеленому цвету, кодируются семейством родственных генов,

Редкая Х-сцепленная рецессивная голубая монохромия или неполная ахроматопсия может быть обусловлена
Редкая Х-сцепленная рецессивная голубая монохромия или неполная ахроматопсия может быть обусловлена

Дихромное цветовое восприятие относится к тяжелым дефектам зрения, при которых используются
Дихромное цветовое восприятие относится к тяжелым дефектам зрения, при которых используются

Аномальная трихромасия, или монохромазия голубых колбочек, протекающая в общем случае гораздо
Аномальная трихромасия, или монохромазия голубых колбочек, протекающая в общем случае гораздо

В каждом из этих двух случаев болезнь обусловлена соответственно мутациями в
В каждом из этих двух случаев болезнь обусловлена соответственно мутациями в

Итак, успешная идентификация целого ряда генов, ответственных за наследственные формы ретинопатий,
Итак, успешная идентификация целого ряда генов, ответственных за наследственные формы ретинопатий,

Основным визуальным пигментом палочек является родопсин.
Абсорбция фотона родопсином приводит к
Основным визуальным пигментом палочек является родопсин. Абсорбция фотона родопсином приводит к

В фоторецепторах активируемый светом фотопигмент взаимодействует с трансдуцином — G-белком, стимулирующим
В фоторецепторах активируемый светом фотопигмент взаимодействует с трансдуцином — G-белком, стимулирующим

Важнейшим звеном фототрансдукции является ретиноидный цикл зрения.
Опсины ковалентно связаны с
Важнейшим звеном фототрансдукции является ретиноидный цикл зрения. Опсины ковалентно связаны с

Очищение фоторецепторов от транс-ретиноля происходит с помощью специфического АТФ-связывающего трансмембранного транспортера,
Очищение фоторецепторов от транс-ретиноля происходит с помощью специфического АТФ-связывающего трансмембранного транспортера,

В нейрональной сети сетчатки свет гиперполяризует фоторецепторы и уменьшает выброс
глютаматного
В нейрональной сети сетчатки свет гиперполяризует фоторецепторы и уменьшает выброс глютаматного

Важнейшую роль в фототрансдукции играет синаптическая передача светового сигнала, в реализации
Важнейшую роль в фототрансдукции играет синаптическая передача светового сигнала, в реализации

Но не только нарушение фототрансдукции приводит к развитию наследственных ретинопатий
Но не только нарушение фототрансдукции приводит к развитию наследственных ретинопатий

Наиболее частые формы врожденного амавроза Лебера и Х-сцепленного пигментного ретинита
Наиболее частые формы врожденного амавроза Лебера и Х-сцепленного пигментного ретинита

К различным вариантам дистрофии колбочек и палочек, пигментного ретинита и
К различным вариантам дистрофии колбочек и палочек, пигментного ретинита и

Атрофия зрительного нерва Лебера является классическим примером митохондриальных заболеваний. Клинически болезнь
Атрофия зрительного нерва Лебера является классическим примером митохондриальных заболеваний. Клинически болезнь
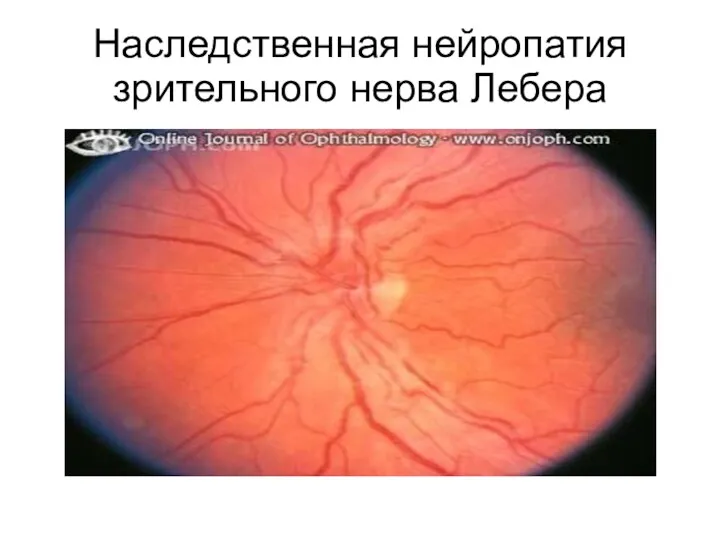
Наследственная нейропатия зрительного нерва Лебера
Наследственная нейропатия зрительного нерва Лебера

Патологические изменения основных структур глазного яблока в большей степени ассоциированы с
Патологические изменения основных структур глазного яблока в большей степени ассоциированы с

Мутации в генах коллагенов, протеогликанов, микрофибриллярных белков или гликопротеинов внеклеточного матрикса
Мутации в генах коллагенов, протеогликанов, микрофибриллярных белков или гликопротеинов внеклеточного матрикса

Мажорный хрящевой коллаген II типа составляет основу стекловидного тела, поэтому его
Мажорный хрящевой коллаген II типа составляет основу стекловидного тела, поэтому его

Её основными клиническими проявлениями является дегенерация стекловидного тела и самопроизвольная отслойка
Её основными клиническими проявлениями является дегенерация стекловидного тела и самопроизвольная отслойка

Наследственные дефекты в коллагене VIII типа, участвующем в образовании десцеметовой мембраны,
Наследственные дефекты в коллагене VIII типа, участвующем в образовании десцеметовой мембраны,

Эпителиально-эндотелиальная дистрофия роговицы Фукса
(буллезная кератопатия, отек стромы)
Эпителиально-эндотелиальная дистрофия роговицы Фукса
(буллезная кератопатия, отек стромы)
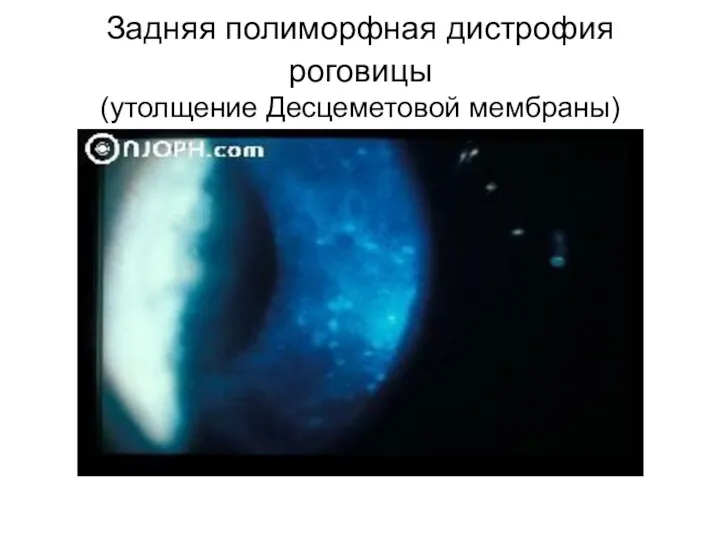
Задняя полиморфная дистрофия роговицы
(утолщение Десцеметовой мембраны)
Задняя полиморфная дистрофия роговицы
(утолщение Десцеметовой мембраны)

Редкий аутосомно-рецессивный синдром Кноблоха (витреоретинальная дегенерация, сочетающаяся с отслоением сетчатки, макулярными
Редкий аутосомно-рецессивный синдром Кноблоха (витреоретинальная дегенерация, сочетающаяся с отслоением сетчатки, макулярными

Аутосомно-доминантная витреоретинальная дегенерация с поздним началом
обусловлена мутациями в гене C1QTNF5,
Аутосомно-доминантная витреоретинальная дегенерация с поздним началом обусловлена мутациями в гене C1QTNF5,

Целый ряд изолированных офтальмопатий обусловлен наследственными дефектами протеогликанов, участвующих в обеспечении
Целый ряд изолированных офтальмопатий обусловлен наследственными дефектами протеогликанов, участвующих в обеспечении

Ранее мы уже обсуждали роль никталопина в развитии одной из генетических
Ранее мы уже обсуждали роль никталопина в развитии одной из генетических

Плоская роговица глаза
(роговица уплощена,тонкая, передняя камера мелкая, исследование при помощи
Плоская роговица глаза (роговица уплощена,тонкая, передняя камера мелкая, исследование при помощи

Кератокан и другие малые кератансульфат-протеогликаны, в первую очередь, люмикан участвуют в
Кератокан и другие малые кератансульфат-протеогликаны, в первую очередь, люмикан участвуют в

Одной из важнейших причин развития наследственных офтальмопатий являются нарушения морфогенеза органа
Одной из важнейших причин развития наследственных офтальмопатий являются нарушения морфогенеза органа

Характерным свойством этой группы заболеваний является огромный клинический полиморфизм, то есть
Характерным свойством этой группы заболеваний является огромный клинический полиморфизм, то есть

Так, не менее 6 аллельных вариантов аутосомно-доминантной дистрофии роговицы обусловлены мутациями
Так, не менее 6 аллельных вариантов аутосомно-доминантной дистрофии роговицы обусловлены мутациями

Это медленно прогрессирующие гранулярные дистрофии I (Гренува) и II (Авеллино) типов,
Это медленно прогрессирующие гранулярные дистрофии I (Гренува) и II (Авеллино) типов,
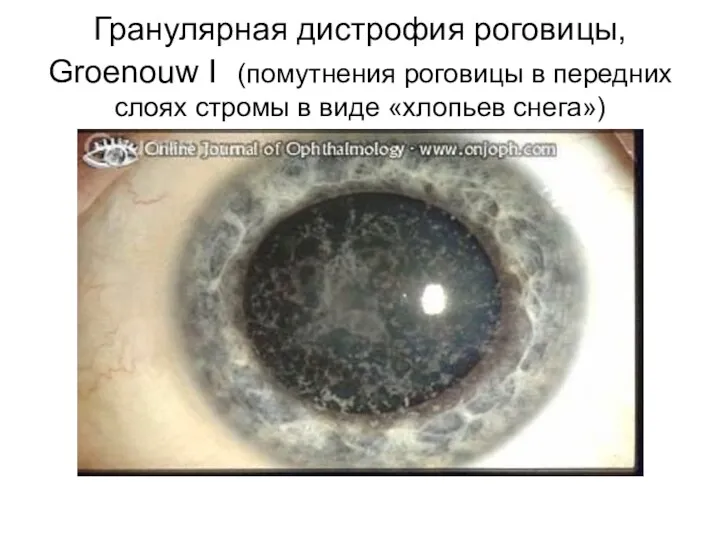
Гранулярная дистрофия роговицы, Groenouw I (помутнения роговицы в передних слоях стромы
Гранулярная дистрофия роговицы, Groenouw I (помутнения роговицы в передних слоях стромы
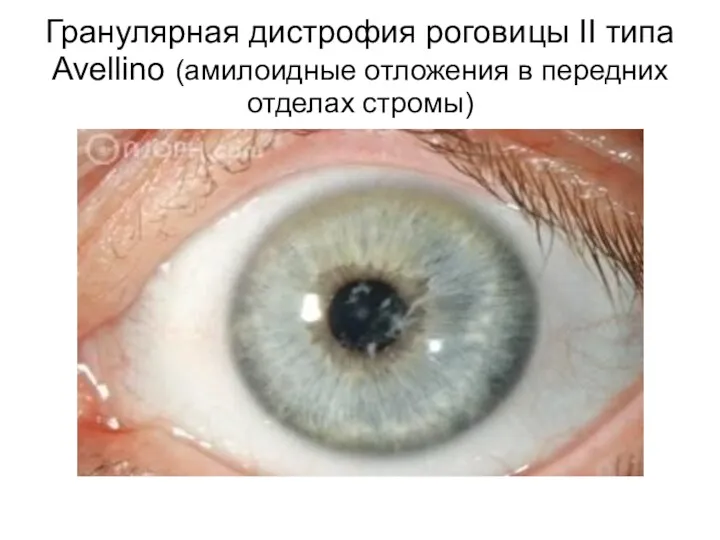
Гранулярная дистрофия роговицы II типа Avellino (амилоидные отложения в передних отделах
Гранулярная дистрофия роговицы II типа Avellino (амилоидные отложения в передних отделах

Дистрофия роговицы Reis-Bückler
(помутнения, расположенные субэпителиально и на боуменовой мембране
Дистрофия роговицы Reis-Bückler
(помутнения, расположенные субэпителиально и на боуменовой мембране
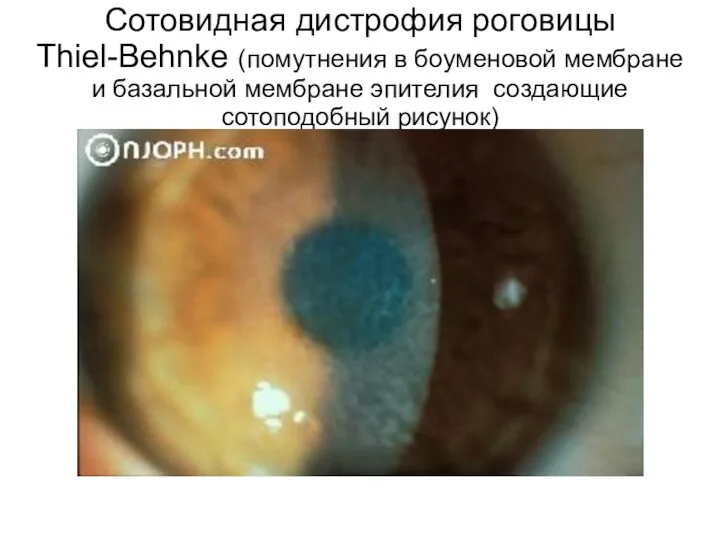
Сотовидная дистрофия роговицы Thiel-Behnke (помутнения в боуменовой мембране и базальной мембране
Сотовидная дистрофия роговицы Thiel-Behnke (помутнения в боуменовой мембране и базальной мембране
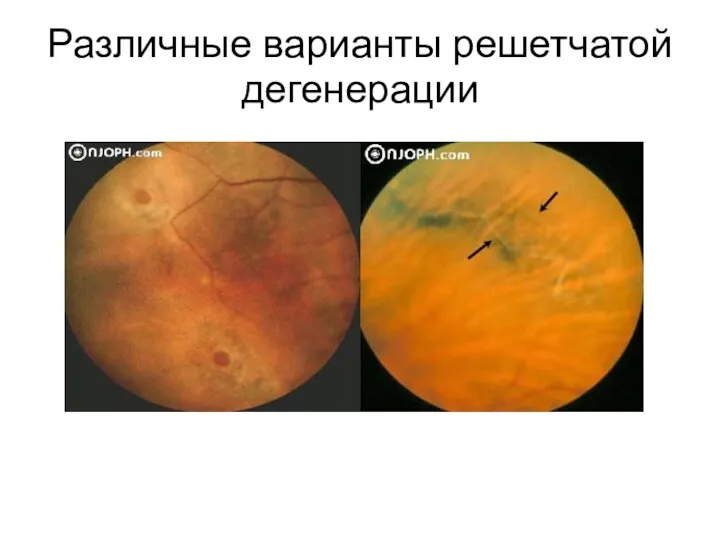
Различные варианты решетчатой дегенерации
Различные варианты решетчатой дегенерации

Однако наибольшее количество изолированных наследственных офтальмопатий связаны с дефектами транскрипционной системы
Однако наибольшее количество изолированных наследственных офтальмопатий связаны с дефектами транскрипционной системы

Два генетических варианта полиморфной задней дистрофии роговицы глаза 3 и 1
Два генетических варианта полиморфной задней дистрофии роговицы глаза 3 и 1
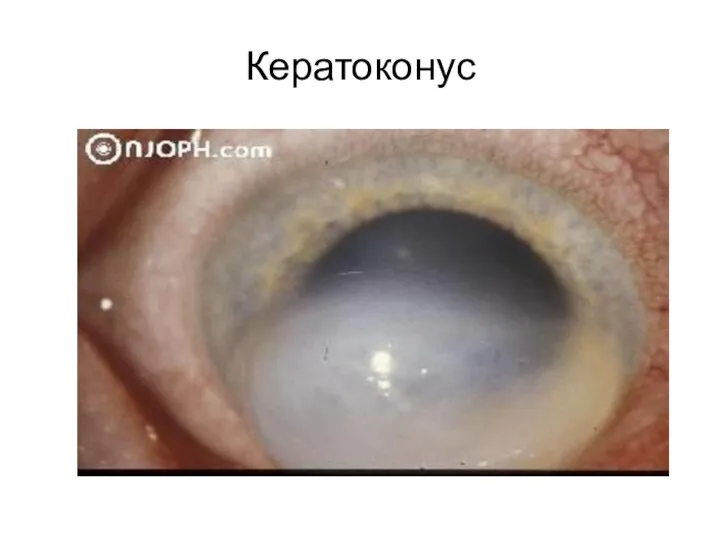
Кератоконус
Кератоконус

Не менее 10 аллельных вариантов аутосомно-доминантных болезней глаз связаны с мутациями
Не менее 10 аллельных вариантов аутосомно-доминантных болезней глаз связаны с мутациями

Это частичная или полная аниридия, аномалии Петерса (недоразвитие стромы роговицы, синехии
Это частичная или полная аниридия, аномалии Петерса (недоразвитие стромы роговицы, синехии
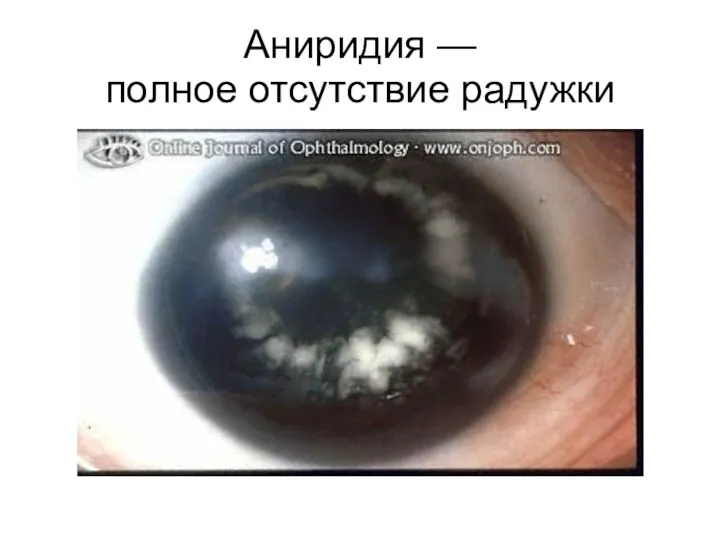
Аниридия —
полное отсутствие радужки
Аниридия —
полное отсутствие радужки

Аниридия со вторичной врожденной глаукомой
Аниридия со вторичной врожденной глаукомой
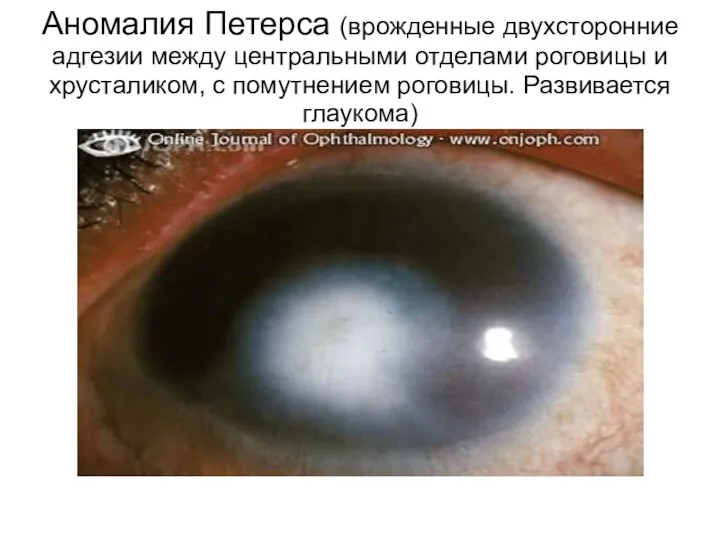
Аномалия Петерса (врожденные двухсторонние адгезии между центральными отделами роговицы и хрусталиком,
Аномалия Петерса (врожденные двухсторонние адгезии между центральными отделами роговицы и хрусталиком,
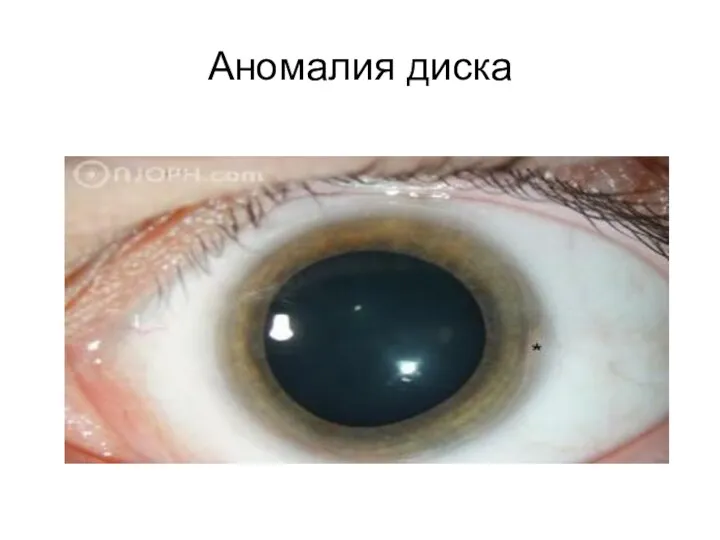
Аномалия диска
Аномалия диска
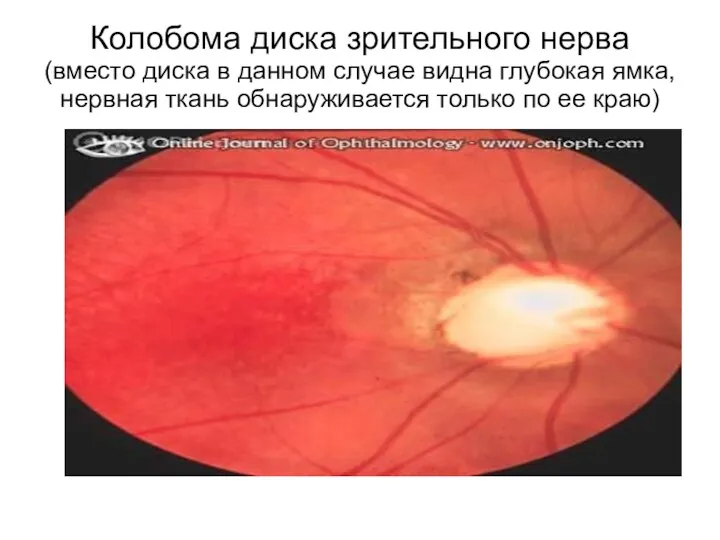
Колобома диска зрительного нерва
(вместо диска в данном случае видна глубокая ямка,
Колобома диска зрительного нерва (вместо диска в данном случае видна глубокая ямка,

Поверхностный точечный билатеральный кератит
Поверхностный точечный билатеральный кератит
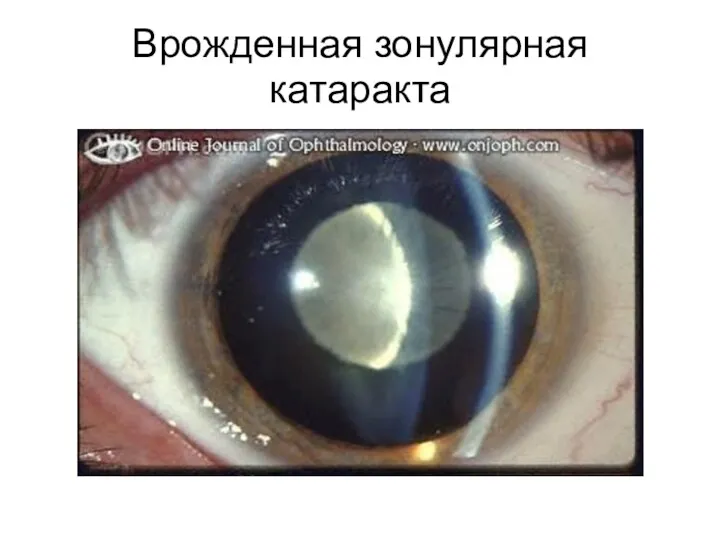
Врожденная зонулярная катаракта
Врожденная зонулярная катаракта

Дефекты развития радужки глаза в сочетании с дегенеративными процессами в роговице,
Дефекты развития радужки глаза в сочетании с дегенеративными процессами в роговице,

Мутации в гене PITX2 идентифицированы при
синдроме Ригера 1 и Аксенфельда-Ригера,
Мутации в гене PITX2 идентифицированы при синдроме Ригера 1 и Аксенфельда-Ригера,
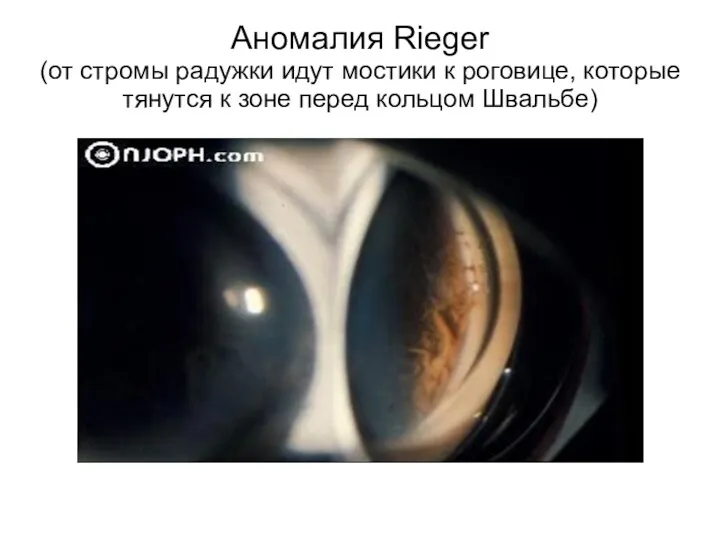
Аномалия Rieger
(от стромы радужки идут мостики к роговице, которые тянутся к
Аномалия Rieger (от стромы радужки идут мостики к роговице, которые тянутся к
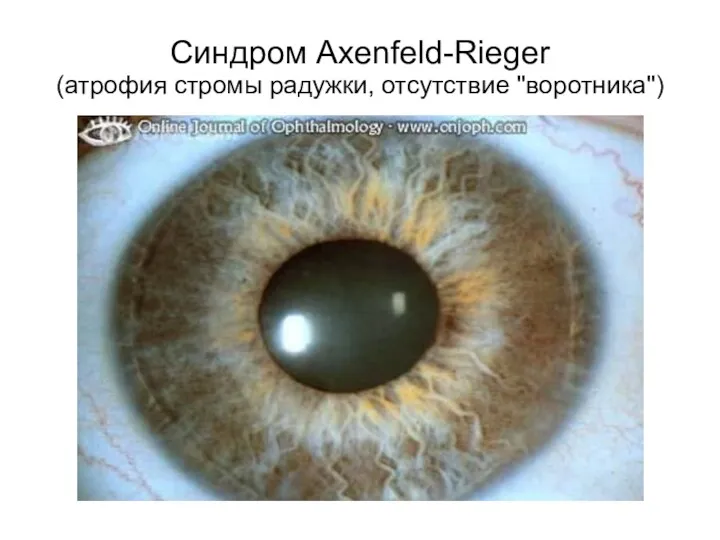
Синдром Axenfeld-Rieger
(атрофия стромы радужки, отсутствие "воротника")
Синдром Axenfeld-Rieger
(атрофия стромы радужки, отсутствие "воротника")

Мезодермальная дисгенезия роговицы и радужки
Мезодермальная дисгенезия роговицы и радужки

Мутации в гене FOXC2 обнаружены при двух аллельных вариантах лимфедемы, сочетающейся
Мутации в гене FOXC2 обнаружены при двух аллельных вариантах лимфедемы, сочетающейся
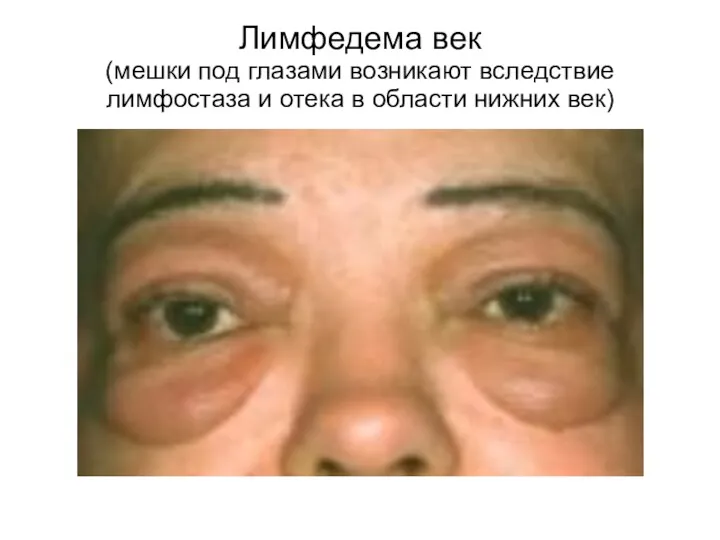
Лимфедема век
(мешки под глазами возникают вследствие лимфостаза и отека в области
Лимфедема век (мешки под глазами возникают вследствие лимфостаза и отека в области

Мутации в гене транскрипционного фактора Sox2, избирательно экспрессирующегося в нервной системе,
Мутации в гене транскрипционного фактора Sox2, избирательно экспрессирующегося в нервной системе,
 Temporary fillings
Temporary fillings Диагностика и лечение инфаркта миокарда
Диагностика и лечение инфаркта миокарда Пиодермии. Определение
Пиодермии. Определение Ана сүтімен қоректендірудің маңызы
Ана сүтімен қоректендірудің маңызы Сосудистый шов
Сосудистый шов Иммунология даму тарихы. Иммунитет теориясы
Иммунология даму тарихы. Иммунитет теориясы Жедел гастрит
Жедел гастрит Принципы диагностики и лечения инфекционных заболеваний
Принципы диагностики и лечения инфекционных заболеваний железа
железа Острые эмболии и тромбозы магистральных артерий конечностей. Консервативное лечение. Показания и виды оперативных вмешательств
Острые эмболии и тромбозы магистральных артерий конечностей. Консервативное лечение. Показания и виды оперативных вмешательств Огнестрельные ранения. Хирургическая обработка огнестрельных ран
Огнестрельные ранения. Хирургическая обработка огнестрельных ран Шок. Патофизиология и принципы интенсивной терапии
Шок. Патофизиология и принципы интенсивной терапии Клинический случай
Клинический случай Вирусные дерматозы
Вирусные дерматозы Проводящая система сердца. ЭКГ
Проводящая система сердца. ЭКГ Особенности обеспечения проходимости дыхательных путей у детей. Интубация трахеи
Особенности обеспечения проходимости дыхательных путей у детей. Интубация трахеи Деваскурялизация матки при применении компрессионного шва по B-Linch
Деваскурялизация матки при применении компрессионного шва по B-Linch Гигиенические требования к выбору и планировке больничного участка. Системы строительства больниц, их преимущества и недостатки
Гигиенические требования к выбору и планировке больничного участка. Системы строительства больниц, их преимущества и недостатки Общая фармакология. Введение в фармакологию
Общая фармакология. Введение в фармакологию Боткин Сергей Петрович
Боткин Сергей Петрович Иық буынының жарақаттары
Иық буынының жарақаттары Респираторный дистресс-синдром взрослых
Респираторный дистресс-синдром взрослых Врождённые пороки развития женской половой системы
Врождённые пороки развития женской половой системы Анатомо-физиологические особенности строения полости рта новорожденного
Анатомо-физиологические особенности строения полости рта новорожденного Дифференциальный диагноз анемий
Дифференциальный диагноз анемий Сухожильный шов
Сухожильный шов Клинико-экономические исследования
Клинико-экономические исследования Дифференциальный диагноз суставного синдрома (один день из жизни врача общей практики)
Дифференциальный диагноз суставного синдрома (один день из жизни врача общей практики)