- Наследственный нефрит

Содержание
- 2. Наследственный нефрит (Синдром Альпорта) - наследственное заболевание, характеризующееся поражением почек с изменениями мочевого осадка ( гематурия
- 3. В 1927 г. A Alport впервые выявил тугоухость у нескольких родственников с гематурией В 1972 г.
- 4. Эпидемиология Распространенность синдрома Альпорта составляет 1 случай на 5000 населения. В Германии синдром Альпорта диагностируется при
- 5. Генетическая основа болезни - мутация в гене а-5 цепи коллагена IV типа. Этот тип универсален для
- 6. Выделяют три варианта наследственного нефрита I вариант - клинически проявляется нефритом с гематурией, тугоухостью и поражением
- 7. II вариант- клинически проявляется нефритом с гематурией без тугоухости. Течение нефрита прогрессирующее с развитием ХПН. Тип
- 8. III вариант - доброкачественная семейная гематурия. Течение благоприятное, хроническая почечная недостаточность не развивается. Тип наследования -
- 9. Синдром Альпорта - наследственный нефрит с поражением слуха. В основе лежит сочетанный дефект структуры колагена базальной
- 10. Коллаген IV типа, входящий в состав гломерулярной базальной мембраны, состоит в основном из двух а1-цепей (IV)
- 11. Структура коллагена IV типа при нормальных и измененных альфа-цепях коллагена: 1 – норма. Все три альфа-цепи
- 14. Морфологически при электронной микроскопии выявляется истончение и расслоение гломерулярных базальных мембран (особенно lamina densa) и наличие
- 15. Полиморфизм ультраструктуры БМ клубочкового капилляра. Участки различной толщины, потеря трехслойности, расслоение и дезорганизация плотной пластины (Lamina
- 16. Симптомы включают в себя: Аномальный цвет мочи, Отёки, Кровь в моче, Снижение или потеря зрения (чаще
- 17. Клиническая картина синдрома Альпорта, регулярно повторяющаяся в семье, обычно соответствует какому-либо фенотипу, хотя выраженность симптомов может
- 18. Фрагмент родословной семьи С. Диагноз: синдром Альпорта
- 19. В начальной стадии болезни самочувствие ребенка страдает мало, характерной особенностью является упорство и стойкость мочевого синдрома:
- 20. В дальнейшем происходит нарушение парциальных функций почек, ухудшение общего состояния больного: появляются интоксикация, мышечная слабость, артериальная
- 21. Для наследственного нефрита характерна стадийность течения болезни: сначала латентная стадия или скрытых клинических симптомов, проявляющаяся минимальными
- 22. Диагностика синдрома Альпорта. Предложены следующие критерии: гематурия или смерть от хронической почечной недостаточности в семейном анамнезе;
- 23. Диагноз синдрома Альпорта считается правомочным в случаях обнаружения у больного 3 из 5 типичных признаков: наличие
- 24. ДИАГНОЗ Важно составление родословной и обнаружение лиц как с сочетанием нефрита и глухоты, так и с
- 25. Дифференциальная диагностика Затруднен дифференциальный диагноз синдрома Альпорта с болезнью тонких базальных мембран, для которой характерны аутосомно-доминантное
- 26. Нефропатия и тугоухость при синдроме Branchio-Oto-Renal сочетается с рудиментарными остатками жаберных щелей . Для синдрома Макла-Уэльса
- 27. Лечение синдрома Альпорта. Специального лечения не существует. Лечение сводится к устранению тех или иных симптомов болезни,
- 28. Исследование Медико-генетического центр РАМН было проведено молекулярно-генетический анализ 16 детям, определил наличие мутации в гене COL4A5
- 29. Среди наблюдаемых больных у 4 мальчиков терминальная стадия ХПН наступила в 14–15 лет, что потребовало проведения
- 31. Скачать презентацию

Наследственный нефрит (Синдром Альпорта) - наследственное заболевание, характеризующееся поражением почек с
Наследственный нефрит (Синдром Альпорта) - наследственное заболевание, характеризующееся поражением почек с

В 1927 г. A Alport впервые выявил тугоухость у нескольких родственников
В 1927 г. A Alport впервые выявил тугоухость у нескольких родственников

Эпидемиология
Распространенность синдрома Альпорта составляет 1 случай на 5000 населения.
В Германии
Эпидемиология
Распространенность синдрома Альпорта составляет 1 случай на 5000 населения.
В Германии

Генетическая основа болезни - мутация в гене а-5 цепи коллагена IV
Генетическая основа болезни - мутация в гене а-5 цепи коллагена IV

Выделяют три варианта наследственного нефрита
I вариант - клинически проявляется нефритом
Выделяют три варианта наследственного нефрита
I вариант - клинически проявляется нефритом

II вариант- клинически проявляется нефритом с гематурией без тугоухости. Течение нефрита
II вариант- клинически проявляется нефритом с гематурией без тугоухости. Течение нефрита

III вариант - доброкачественная семейная гематурия.
Течение благоприятное, хроническая почечная недостаточность
III вариант - доброкачественная семейная гематурия.
Течение благоприятное, хроническая почечная недостаточность

Синдром Альпорта - наследственный нефрит с поражением слуха.
В основе лежит сочетанный дефект
Синдром Альпорта - наследственный нефрит с поражением слуха.
В основе лежит сочетанный дефект

Коллаген IV типа, входящий в состав гломерулярной базальной мембраны, состоит в
Коллаген IV типа, входящий в состав гломерулярной базальной мембраны, состоит в
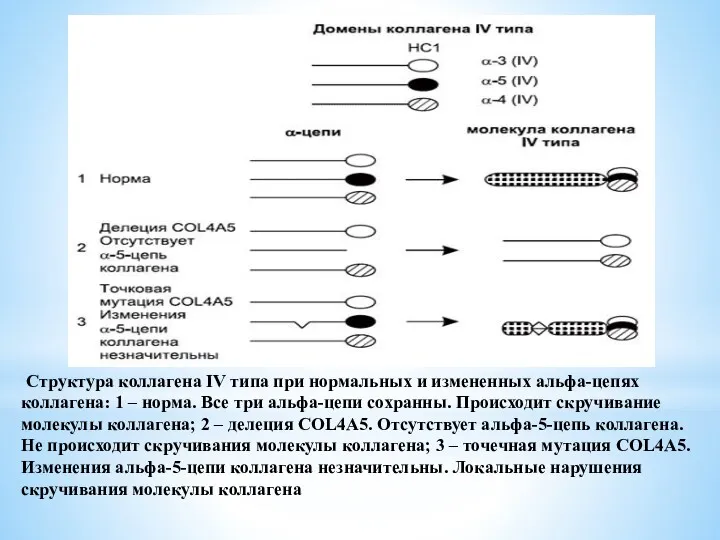
Структура коллагена IV типа при нормальных и измененных альфа-цепях коллагена:
Структура коллагена IV типа при нормальных и измененных альфа-цепях коллагена:

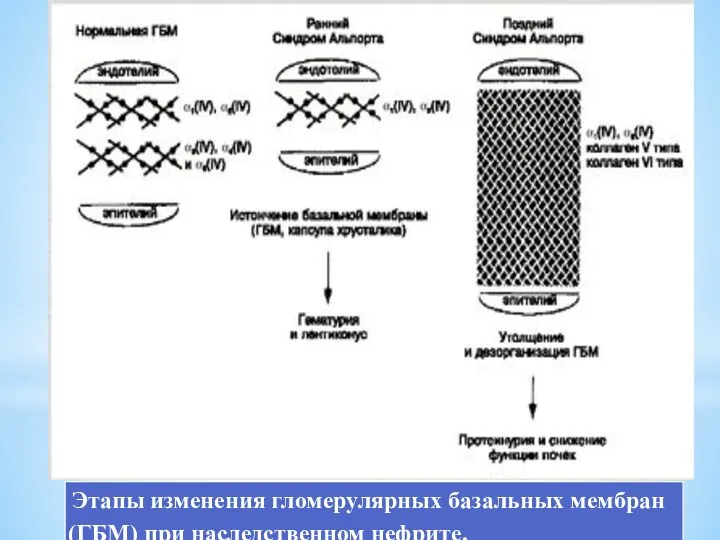

Морфологически при электронной микроскопии выявляется истончение и расслоение гломерулярных базальных мембран
Морфологически при электронной микроскопии выявляется истончение и расслоение гломерулярных базальных мембран
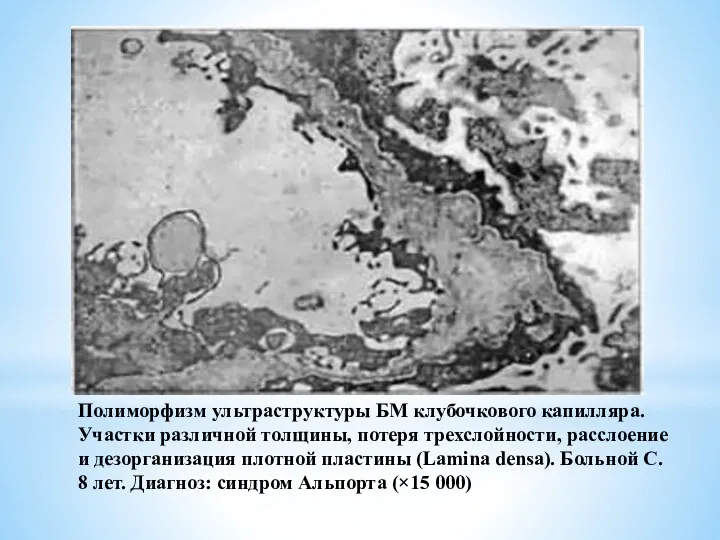
Полиморфизм ультраструктуры БМ клубочкового капилляра. Участки различной толщины, потеря трехслойности, расслоение
Полиморфизм ультраструктуры БМ клубочкового капилляра. Участки различной толщины, потеря трехслойности, расслоение
Симптомы включают в себя:
Аномальный цвет мочи,
Отёки,
Кровь в моче,
Снижение или потеря зрения
Симптомы включают в себя:
Аномальный цвет мочи,
Отёки,
Кровь в моче,
Снижение или потеря зрения

Клиническая картина синдрома Альпорта, регулярно повторяющаяся в семье, обычно соответствует какому-либо
Клиническая картина синдрома Альпорта, регулярно повторяющаяся в семье, обычно соответствует какому-либо
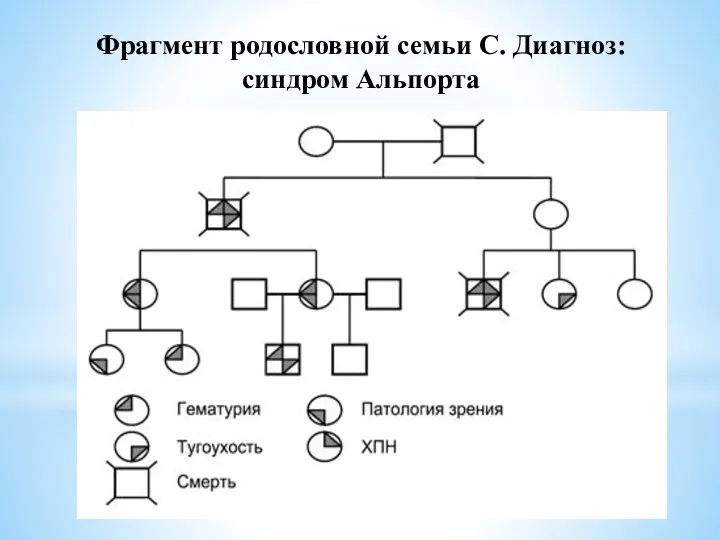
Фрагмент родословной семьи С. Диагноз: синдром Альпорта
Фрагмент родословной семьи С. Диагноз: синдром Альпорта

В начальной стадии болезни самочувствие ребенка страдает мало, характерной особенностью является
В начальной стадии болезни самочувствие ребенка страдает мало, характерной особенностью является

В дальнейшем происходит нарушение парциальных функций почек, ухудшение общего состояния больного:
В дальнейшем происходит нарушение парциальных функций почек, ухудшение общего состояния больного:

Для наследственного нефрита характерна стадийность течения болезни: сначала латентная стадия или
Для наследственного нефрита характерна стадийность течения болезни: сначала латентная стадия или

Диагностика синдрома Альпорта.
Предложены следующие критерии:
гематурия или смерть от хронической почечной недостаточности
Диагностика синдрома Альпорта.
Предложены следующие критерии:
гематурия или смерть от хронической почечной недостаточности

Диагноз синдрома Альпорта считается правомочным в случаях обнаружения у больного 3
Диагноз синдрома Альпорта считается правомочным в случаях обнаружения у больного 3

ДИАГНОЗ
Важно составление родословной и обнаружение лиц как с сочетанием нефрита и
ДИАГНОЗ
Важно составление родословной и обнаружение лиц как с сочетанием нефрита и

Дифференциальная диагностика
Затруднен дифференциальный диагноз синдрома Альпорта с болезнью тонких базальных мембран,
Дифференциальная диагностика
Затруднен дифференциальный диагноз синдрома Альпорта с болезнью тонких базальных мембран,

Нефропатия и тугоухость при синдроме Branchio-Oto-Renal сочетается с рудиментарными остатками жаберных
Нефропатия и тугоухость при синдроме Branchio-Oto-Renal сочетается с рудиментарными остатками жаберных

Лечение синдрома Альпорта.
Специального лечения не существует. Лечение сводится к устранению тех
Лечение синдрома Альпорта.
Специального лечения не существует. Лечение сводится к устранению тех
Исследование
Медико-генетического центр РАМН было проведено молекулярно-генетический анализ 16 детям, определил наличие
Исследование
Медико-генетического центр РАМН было проведено молекулярно-генетический анализ 16 детям, определил наличие

Среди наблюдаемых больных у 4 мальчиков терминальная стадия ХПН наступила в
Среди наблюдаемых больных у 4 мальчиков терминальная стадия ХПН наступила в
 Физиология дыхания
Физиология дыхания Кожное заболевание чесотка
Кожное заболевание чесотка Воспалительные заболевания слуховой трубы
Воспалительные заболевания слуховой трубы История болезни
История болезни Антибиотики. Определение
Антибиотики. Определение Теоретическая основа здравоохранения и фармации
Теоретическая основа здравоохранения и фармации Эндокринология. Общая эндокринология. Частная эндокринология
Эндокринология. Общая эндокринология. Частная эндокринология Умирание, смерть и трупные изменения
Умирание, смерть и трупные изменения Перекрестный прикус
Перекрестный прикус Шок – собирательный термин, обозначающий критическое состояние
Шок – собирательный термин, обозначающий критическое состояние Профилактика ранней беременности
Профилактика ранней беременности Роль и место препаратов сульфонилмочевины в терапии сахарного диабета 2 типа
Роль и место препаратов сульфонилмочевины в терапии сахарного диабета 2 типа Общая реакция организма на повреждение
Общая реакция организма на повреждение Общие вопросы стоматологии. Организация работы врача - стоматолога-терапевта
Общие вопросы стоматологии. Организация работы врача - стоматолога-терапевта Канцерогены, как факторы опухолевого роста
Канцерогены, как факторы опухолевого роста Алергія. Типи алергічних реакцій
Алергія. Типи алергічних реакцій Психопатология, психопатия и акцентуация характера
Психопатология, психопатия и акцентуация характера Кешенді медициналық ақпараттық жүйе (КМИС)
Кешенді медициналық ақпараттық жүйе (КМИС) Сахарный диабет первого типа. Определение, распространение, социальное значение. Клиника, диагностика, лечение
Сахарный диабет первого типа. Определение, распространение, социальное значение. Клиника, диагностика, лечение Пародонттың қызметі және
Пародонттың қызметі және Бронхиальная астма, сердечная астма, астма при уремии
Бронхиальная астма, сердечная астма, астма при уремии Сит задачи по ОЖ экзаменац
Сит задачи по ОЖ экзаменац Одонтогенные верхнечелюстные синуситы
Одонтогенные верхнечелюстные синуситы Показания к оперативному лечению. Методы предоперационного обследования
Показания к оперативному лечению. Методы предоперационного обследования Острый коронарный синдром: инфаркт миокарда без подъема ST
Острый коронарный синдром: инфаркт миокарда без подъема ST Острые лейкозы
Острые лейкозы ВИЧ/СПИД
ВИЧ/СПИД Фармакология кардиотонических препаратов
Фармакология кардиотонических препаратов