- Основы клинической ферментологии. Энзимопатология

Содержание
- 2. Энзимопатии заболевания, связанные с нарушением активности определённых ферментов
- 3. Классификация энзимопатий Врождённые Токсические Алиментарные Нейро - эндокринные Структурные Прочие
- 4. Наследственные энзимопатии выявляются в раннем детском возрасте
- 5. Наследственные энзимопатии Классические обусловлены полным выпадением синтеза фермента обусловлены слабостью отдельных звеньев ферментативного процесса Регуляторные Обусловлены
- 6. Токсические энзимопатии Связанные с избирательным угнетением активности одного фермента Связанные со специфическим угнетением синтеза ферментов Связанные
- 7. Алиментарные энзимопатии Вызванные дефицитом витаминов Вызванные дефицитом микроэлементов Вызванные дефицитом белка Вызванные разбалансированностью питания
- 8. Классические врождённые энзимопатии Схема ферментативного блока Необычные (токсичные) метаболиты Субстрат Продукты реакции Фермент не образуются отсутствует
- 9. Проявления ферментативного блока Снижение концентрации продукта реакции Повышение концентрации субстрата (или его предшественника) При наличии альтернативных
- 10. Энзимопатии обмена аминокислот
- 11. Нарушения обмена фенилаланина Фенилкетонурия (фенилпировиноградная олигофрения) блок фенилаланинмонооксигеназы в печени, почках, поджелудочной железе Фенилаланин тирозин Фенилпируват
- 12. ФКУ Ранние симптомы: повышенная возбудимость, двигательная гиперактивность, экземоподобная сыпь, запах плесени от пота и мочи. До
- 13. Нарушение обмена тирозина Тирозинемии (тирозинозы) Тирозин Трансаминаза (II тип) Парагидроксифенилпируват Оксидаза ( у новорожденных) Фумарилацетат Гидролаза
- 14. Транзиторная тирозинемия новорожденных – дефект оксидазы параоксифенилпирувата Параоксифенилпируват гомогентизиновая к-та Встречается чаще других нарушений обмена аминокислот
- 15. Наследственная тирозинемия I типа- блок гидроксилазы фумарилацетоуксусной кислоты Фумарилацетоацетат фумарат + ацетоацетат Острое проявление в возрасте
- 16. Алкаптонурия- нарушение обмена тирозина Алкаптонурия (черная моча) Блок оксидазы гомогентизиновой кислоты Тирозин гомогентизиновая кислота фумарилацетоуксусная кислота
- 17. Алкаптонурия Симптомы: окрашивание мочи в чёрный цвет, другие клинические проявления в детском возрасте отсутствуют. В более
- 18. Нарушение обмена тирозина Альбинизм блок ферментов синтеза меланинов Тирозин ДОФА Меланины
- 19. Нарушения синтеза мочевины карбамоилфосфатсинтетаза NH3 карбамоилфосфат карбамоилорнитинтрансфераза гипераммониемия цитруллин цитруллинемия аргининсукцинатсинтетаза аргининсукцинат аргининсукцинатурия аргининсукцинатлиаза аргинин аргининемия
- 20. Гипераммониемия I типа- блок карбамоилфосфатсинтетазы Аммиак карбамоилфосфат Молниеносное течение (в период новорожденности после кормления молоком) повышенная
- 21. Аргининсукцинатурния- блок аргининсукцинатлиазы Аргининсукцинат аргинин + фумарат Наиболее частое заболевание в цикле мочевины При остром течении
- 22. Общие принципы лечения нарушений в цикле мочевины Ограничение белка до минимально возможного уровня. Клизмы для очистки
- 23. Нарушение обмена серосодержащих аминокислот Метионин S аденозилметионин ТГФК [СН3] В12 [СН3] Гомоцистеин S аденозилденозилгомоцистеин серин цистатион
- 24. Гомоцистинурия- дефект цистатион-β- синтетазы Метионин цистатион цистеин Симптомы: поражения глаз, скелета, сосудистой системы, мозга. Вывих хрусталика
- 25. Цистиноз- блок лизосомных ферментов обмена цистеина Симптомы появляются после 6 месяцев жизни: полиурия, полидипсия, лихорадка, гипотрофия,
- 26. Энзимопатии углеводного обмена
- 27. Врождённая недостаточность лактазы кишечника Лактоза глюкоза + галактоза Клинические симптомы проявляются у новорожденных после начала кормления
- 28. Недостаточность сахаразы-изомальтазы кишечника Сахароза глюкоза + фруктоза Симптомы выявляются при введении прикорма, содержащего сахарозу: хроническая диарея,
- 29. Нарушение тканевого обмена галактозы(галактозо-1-фосфатуридилтрансфераза) галактокиназа гексозо-1-фосфатуридилтрансфераза Галактоза Галактозо-1-фосфат глюкозо-1-фосфат Галактоземия недостаточность гексозо-1-фосфатуридилтрансферазы Симптомы возникают в первые
- 30. Фруктоземия- недостаточность фруктозо-1-фосфатальдолазы в тканях фруктозо-1-фосфатальдолаза Фруктоза фруктозо-1-фосфат 2 триозы Фосфат включается во фруктозо-1-фосфат гипофосфатемия, снижение
- 31. Гликогеновые болезни
- 32. Гликогеноз I типа- болезнь Гирке дефект глюкозо-6-фосфатазы Глюкозо-6-фосфат глюкоза Глюкозо-6-фосфат в гликолиз лактат торможение экскреции уратов
- 33. Гликогеновая болезнь 1 типа
- 34. Гликогеноз II типа –болезнь Помпе дефект лизосомной альфа-1,4- глюкозидазы гликоген глюкоза Симптомы проявляются в начале 1
- 35. Гликогеноз III типа – болезнь Кори, болезнь Форбса -дефект амило-1,6- глюкозидазы Страдает расщепление ответвлений в гликогене
- 36. Гликогеноз V типа –болезнь Мак-Ардла недостаточность миофосфорилазы Гликоген + Н3РО4 Глюкозо-1 фосфат Мышечные боли даже после
- 37. Выделяют 14 типов мукополисахаридозов, из них наиболее часто встречаются типы I и II. Мукополисахаридоз I типа
- 38. Болезнь Гурлера Идуронидаза Дерматансульфаты Гепарансульфаты Гепарансульфаты Болезнь Гюнтера Идуронатсульфатаза Дерматансульфаты Болезнь Моркио Хондроитин-6-сульфатаза Кератансульфаты Болезнь Слая
- 39. Синдром Гурлер: типичные внешние проявления. При синдроме Гурлера снижена активность лизосомального фермента альфа-L-идуронидазы, которая катализирует гидролиз
- 40. Идуронидаза
- 41. Синдром Гурлера- дефект α-идуронидазы, Грубая задержка психомоторного развития, мегалоцефалия, короткая шея, запавшее переносье, макроглоссия, пупочная грыжа,
- 42. К МПС II относится синдром Хантера. Он представляет собой рецессивное наследственное заболевание, вызванное недостаточным уровнем в
- 43. Сульфатаза
- 44. Синдром Моркио Дефект галактозо-6-сульфатазы, нарушение распада кератансульфатов. (Галактоза- N-ацетилглюкозамин-сульфат) Симптомы: преимущественное поражение скелета, Х-образное искривление ног,
- 46. Терапия МПС Ферментозаместительная терапия Элапраза Ларонидаза (альдуразим) Трансплантация гемопоэтических стволовых клеток Генотерапия ( с применением вирусных
- 47. Ферментозамещающая терапия мукополисахаридозов
- 48. Энзимопатии липидного обмена
- 49. Энзимопатии липидного обмена Сфинголипидозы Болезнь Гоше (цереброзидоз) недостаточность бета-гликозидазы Церамид- олигосахарид- гексозамин недостаточность гексаминидазы Болезнь Тея
- 50. Структура сфингомиелинов
- 52. Сфингомиелиноз –болезнь Нимана-Пика недостаточность лизосомной сфингомиелиназы Церамид - фосфат- холин Младенческий вариант А до 3 месяцев:
- 53. - манифестация основных симптомов заболевания на первом году жизни; - грубые черты лица; - тугоподвижность суставов;
- 54. тип А тип В
- 55. - манифестация основных симптомов заболевания на первом году жизни; - грубые черты лица; - тугоподвижность суставов;
- 56. ЦЕРЕБРОЗИД
- 57. Распад цереброзидов Дефект фермента вызывает цереброзидоз (болезнь Гоше) β-ГЛИКОЗИДАЗА CH3 | (CH2)12 | CH || CH
- 58. Цереброзидоз –болезнь Гоше недостаточность лизосомальной β-гликозидазы Церамид - галактоза Накапливание цереброзида в ретикулоэндотелиальных клетках печени, селезёнки,
- 59. Ганглиозидоз -болезнь Тея-Сакса недостаточность лизосомной гексаминидазы А и В Церамид - олигосахарид - аминогексоза Младенческая и
- 60. Ферментозамещающая терапия сфинголипидозов
- 61. Аферментозы обмена нуклеотидов болезнь Леш-Нихана Недостаточность гипоксантин-гуанин-фосфорибозилтрансферазы Гуанин + ФРПФ ГМФ мочевая кислота Оротатацидурия – накопление
- 62. Энзимопатии обмена гемоглобина Фавизм (гемолитическая анемия) недостаточность глюкозо-6-фосфатдегидрогеназы Врождённая метгемоглобинопатия – недостаточность НАДН-цитохром В5-редуктазы Порфирии –
- 63. Порфирии- нарушение активности ферментов синтеза гема Клинически выделяют : печёночные порфирии с выраженными неврологическими нарушениями вследствие
- 64. Классификация порфирий
- 65. Нарушения обмена билирубина Синдром Криглера – Найяра (неконьюгированная желтуха) Отсутствие глюкуронилтрансферазы Гипербилирубинемия за счёт свободного (непрямого)
- 66. Транзиторная желтуха новорожденных Усилен гемолиз эритроцитов Снижено содержание альбуминов Снижен захват билирубина печенью Снижена активность глюкуронил
- 67. Митохондриальные болезни Проявляются нарушением тканевого дыхания, миопатией, поражением нервной системы, печени, сердца, нарушением слуха, зрения, функции
- 68. Нарушения обмена жирных кислот Группа тяжёлых наследственных болезней, характеризующихся высокой смертностью, преимущественным поражением цнс, сердца, печени.
- 69. Клинические симптомы нарушения обмена жирных кислот Неонатальная Детская Поздняя формы Энцефалопатия (вялость, сонливость, кома), рвота, кардиомиопатия,
- 70. Биохимические сдвиги при нарушении обмена жирных кислот Не происходит бета-окисления, активируется омега-окисление с накоплением дикарбоновых кислот,
- 71. Диагностика нарушений обмена жирных кислот Определение уровня карнитина Определение экскреции органических кислот Снижение активности ферментов обмена
- 72. Лечение нарушений обмена жирных кислот Диетотерапия (исключение голода, обогащение рациона углеводами при снижении содержания липидов и
- 74. Скачать презентацию

Энзимопатии
заболевания, связанные с нарушением активности определённых ферментов
Энзимопатии
заболевания, связанные с нарушением активности определённых ферментов

Классификация энзимопатий
Врождённые
Токсические
Алиментарные
Нейро - эндокринные
Структурные
Прочие
Классификация энзимопатий
Врождённые
Токсические
Алиментарные
Нейро - эндокринные
Структурные
Прочие
Наследственные энзимопатии
выявляются в раннем детском возрасте
Наследственные энзимопатии
выявляются в раннем детском возрасте
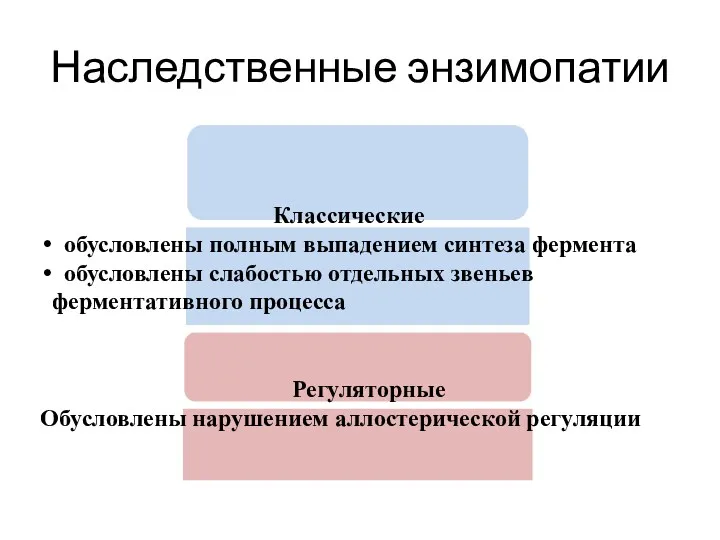
Наследственные энзимопатии
Классические
обусловлены полным выпадением синтеза фермента
обусловлены слабостью отдельных звеньев
Наследственные энзимопатии
Классические
обусловлены полным выпадением синтеза фермента
обусловлены слабостью отдельных звеньев
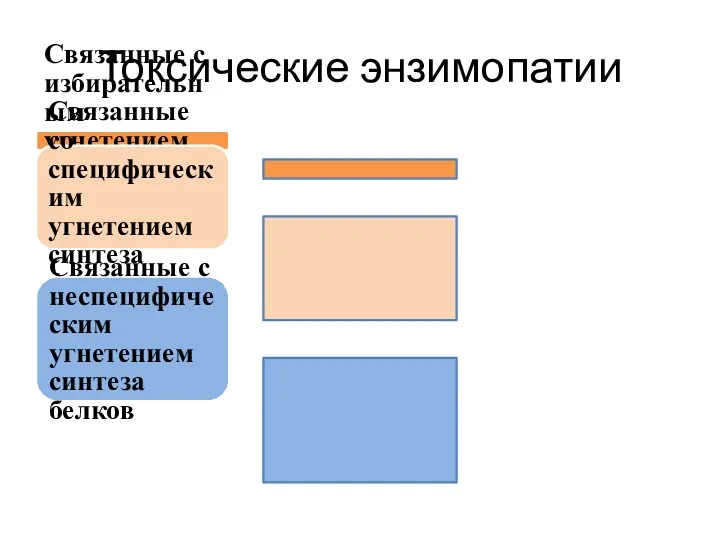
Токсические энзимопатии
Связанные с избирательным угнетением активности одного фермента
Связанные со специфическим угнетением
Токсические энзимопатии
Связанные с избирательным угнетением активности одного фермента
Связанные со специфическим угнетением

Алиментарные
энзимопатии
Вызванные дефицитом
витаминов
Вызванные дефицитом микроэлементов
Вызванные дефицитом
белка
Вызванные разбалансированностью
Алиментарные
энзимопатии
Вызванные дефицитом
витаминов
Вызванные дефицитом микроэлементов
Вызванные дефицитом
белка
Вызванные разбалансированностью
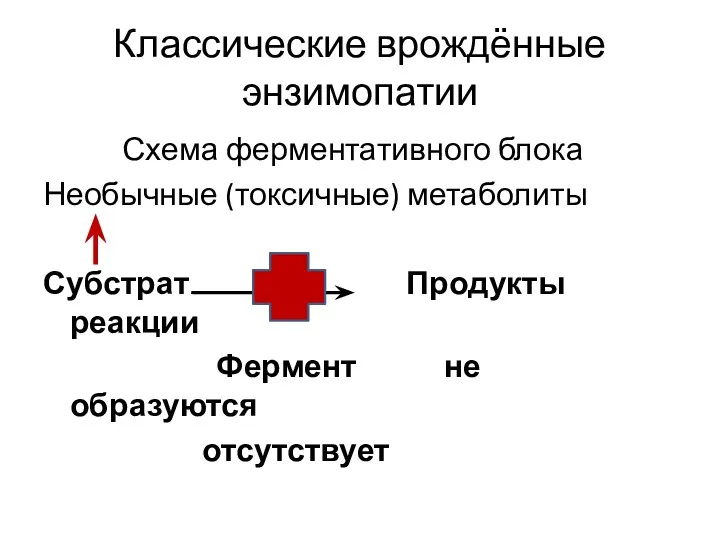
Классические врождённые энзимопатии
Схема ферментативного блока
Необычные (токсичные) метаболиты
Субстрат Продукты реакции
Фермент
Классические врождённые энзимопатии
Схема ферментативного блока
Необычные (токсичные) метаболиты
Субстрат Продукты реакции
Фермент

Проявления ферментативного блока
Снижение концентрации продукта реакции
Повышение концентрации субстрата (или его предшественника)
При
Проявления ферментативного блока
Снижение концентрации продукта реакции
Повышение концентрации субстрата (или его предшественника)
При

Энзимопатии обмена аминокислот
Энзимопатии обмена аминокислот
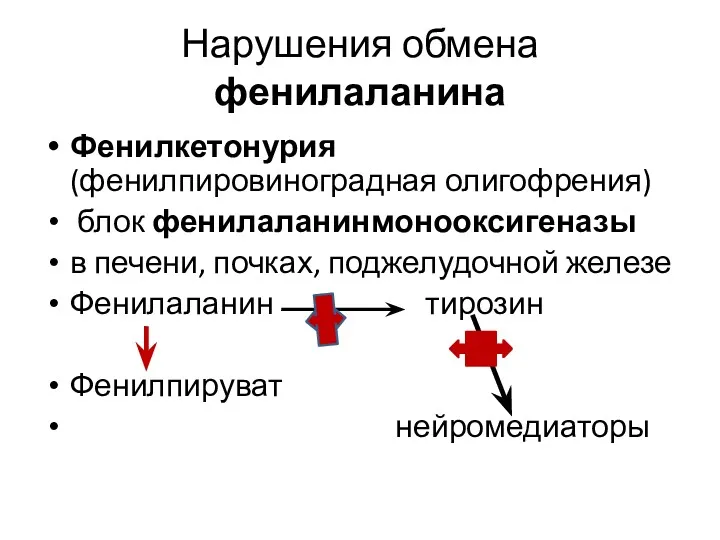
Нарушения обмена фенилаланина
Фенилкетонурия (фенилпировиноградная олигофрения)
блок фенилаланинмонооксигеназы
в печени,
Нарушения обмена фенилаланина
Фенилкетонурия (фенилпировиноградная олигофрения)
блок фенилаланинмонооксигеназы
в печени,

ФКУ
Ранние симптомы: повышенная возбудимость, двигательная гиперактивность, экземоподобная сыпь, запах плесени от
ФКУ
Ранние симптомы: повышенная возбудимость, двигательная гиперактивность, экземоподобная сыпь, запах плесени от

Нарушение обмена тирозина
Тирозинемии (тирозинозы)
Тирозин
Трансаминаза (II тип)
Парагидроксифенилпируват
Оксидаза ( у
Нарушение обмена тирозина
Тирозинемии (тирозинозы)
Тирозин
Трансаминаза (II тип)
Парагидроксифенилпируват
Оксидаза ( у

Транзиторная тирозинемия новорожденных –
дефект оксидазы параоксифенилпирувата
Параоксифенилпируват гомогентизиновая к-та
Встречается чаще других нарушений
Транзиторная тирозинемия новорожденных –
дефект оксидазы параоксифенилпирувата
Параоксифенилпируват гомогентизиновая к-та
Встречается чаще других нарушений

Наследственная тирозинемия I типа-
блок гидроксилазы фумарилацетоуксусной кислоты
Фумарилацетоацетат фумарат + ацетоацетат
Острое проявление
Наследственная тирозинемия I типа-
блок гидроксилазы фумарилацетоуксусной кислоты
Фумарилацетоацетат фумарат + ацетоацетат
Острое проявление
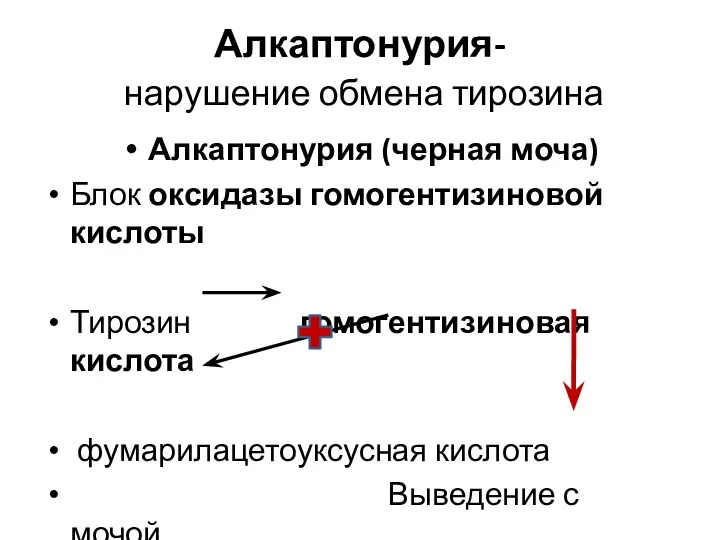
Алкаптонурия-
нарушение обмена тирозина
Алкаптонурия (черная моча)
Блок оксидазы гомогентизиновой кислоты
Тирозин гомогентизиновая кислота
Алкаптонурия-
нарушение обмена тирозина
Алкаптонурия (черная моча)
Блок оксидазы гомогентизиновой кислоты
Тирозин гомогентизиновая кислота

Алкаптонурия
Симптомы: окрашивание мочи в чёрный цвет, другие клинические проявления в детском
Алкаптонурия
Симптомы: окрашивание мочи в чёрный цвет, другие клинические проявления в детском

Нарушение обмена тирозина
Альбинизм
блок ферментов
синтеза меланинов
Тирозин
ДОФА
Меланины
Нарушение обмена тирозина
Альбинизм
блок ферментов
синтеза меланинов
Тирозин
ДОФА
Меланины
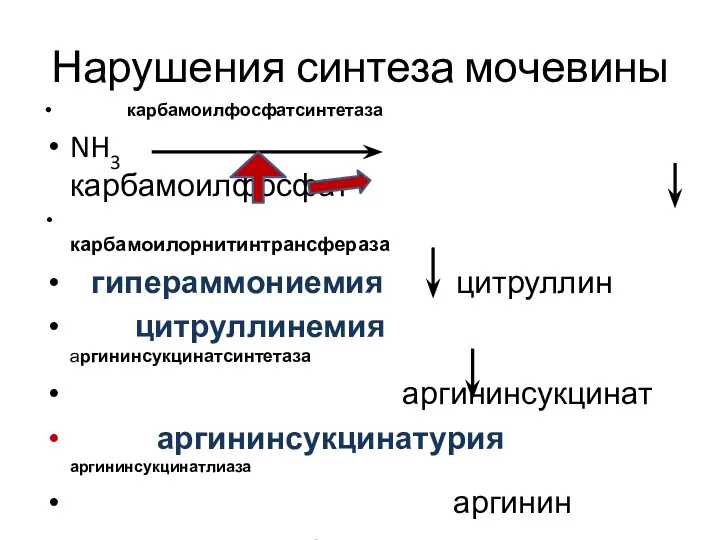
Нарушения синтеза мочевины
карбамоилфосфатсинтетаза
NH3 карбамоилфосфат
карбамоилорнитинтрансфераза
гипераммониемия цитруллин
цитруллинемия аргининсукцинатсинтетаза
аргининсукцинат
Нарушения синтеза мочевины
карбамоилфосфатсинтетаза
NH3 карбамоилфосфат
карбамоилорнитинтрансфераза
гипераммониемия цитруллин
цитруллинемия аргининсукцинатсинтетаза
аргининсукцинат

Гипераммониемия I типа-
блок карбамоилфосфатсинтетазы
Аммиак карбамоилфосфат
Молниеносное течение (в период новорожденности после
Гипераммониемия I типа-
блок карбамоилфосфатсинтетазы
Аммиак карбамоилфосфат
Молниеносное течение (в период новорожденности после

Аргининсукцинатурния-
блок аргининсукцинатлиазы
Аргининсукцинат аргинин + фумарат
Наиболее частое заболевание в цикле мочевины
При остром
Аргининсукцинатурния-
блок аргининсукцинатлиазы
Аргининсукцинат аргинин + фумарат
Наиболее частое заболевание в цикле мочевины
При остром

Общие принципы лечения нарушений в цикле мочевины
Ограничение белка до минимально возможного
Общие принципы лечения нарушений в цикле мочевины
Ограничение белка до минимально возможного
![Нарушение обмена серосодержащих аминокислот Метионин S аденозилметионин ТГФК [СН3] В12](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/253687/slide-22.jpg)
Нарушение обмена серосодержащих аминокислот
Метионин S аденозилметионин
ТГФК
[СН3]
В12 [СН3]
Гомоцистеин S аденозилденозилгомоцистеин
серин
Нарушение обмена серосодержащих аминокислот
Метионин S аденозилметионин
ТГФК
[СН3]
В12 [СН3]
Гомоцистеин S аденозилденозилгомоцистеин
серин

Гомоцистинурия-
дефект цистатион-β- синтетазы
Метионин цистатион цистеин
Симптомы: поражения глаз, скелета, сосудистой системы,
Гомоцистинурия-
дефект цистатион-β- синтетазы
Метионин цистатион цистеин
Симптомы: поражения глаз, скелета, сосудистой системы,

Цистиноз-
блок лизосомных ферментов обмена цистеина
Симптомы появляются после 6 месяцев жизни: полиурия,
Цистиноз-
блок лизосомных ферментов обмена цистеина
Симптомы появляются после 6 месяцев жизни: полиурия,

Энзимопатии углеводного обмена
Энзимопатии углеводного обмена

Врождённая недостаточность лактазы кишечника
Лактоза глюкоза + галактоза
Клинические симптомы проявляются у
Врождённая недостаточность лактазы кишечника
Лактоза глюкоза + галактоза
Клинические симптомы проявляются у

Недостаточность сахаразы-изомальтазы кишечника
Сахароза глюкоза + фруктоза
Симптомы выявляются при введении прикорма,
Недостаточность сахаразы-изомальтазы кишечника
Сахароза глюкоза + фруктоза
Симптомы выявляются при введении прикорма,

Нарушение тканевого обмена галактозы(галактозо-1-фосфатуридилтрансфераза)
галактокиназа гексозо-1-фосфатуридилтрансфераза
Галактоза Галактозо-1-фосфат
глюкозо-1-фосфат
Галактоземия
недостаточность гексозо-1-фосфатуридилтрансферазы
Симптомы возникают в
Нарушение тканевого обмена галактозы(галактозо-1-фосфатуридилтрансфераза)
галактокиназа гексозо-1-фосфатуридилтрансфераза
Галактоза Галактозо-1-фосфат
глюкозо-1-фосфат
Галактоземия
недостаточность гексозо-1-фосфатуридилтрансферазы
Симптомы возникают в

Фруктоземия-
недостаточность фруктозо-1-фосфатальдолазы в тканях
фруктозо-1-фосфатальдолаза
Фруктоза фруктозо-1-фосфат 2 триозы
Фосфат включается во фруктозо-1-фосфат
Фруктоземия-
недостаточность фруктозо-1-фосфатальдолазы в тканях
фруктозо-1-фосфатальдолаза
Фруктоза фруктозо-1-фосфат 2 триозы
Фосфат включается во фруктозо-1-фосфат

Гликогеновые болезни
Гликогеновые болезни

Гликогеноз I типа- болезнь Гирке
дефект глюкозо-6-фосфатазы
Глюкозо-6-фосфат глюкоза
Глюкозо-6-фосфат в гликолиз лактат
Гликогеноз I типа- болезнь Гирке
дефект глюкозо-6-фосфатазы
Глюкозо-6-фосфат глюкоза
Глюкозо-6-фосфат в гликолиз лактат
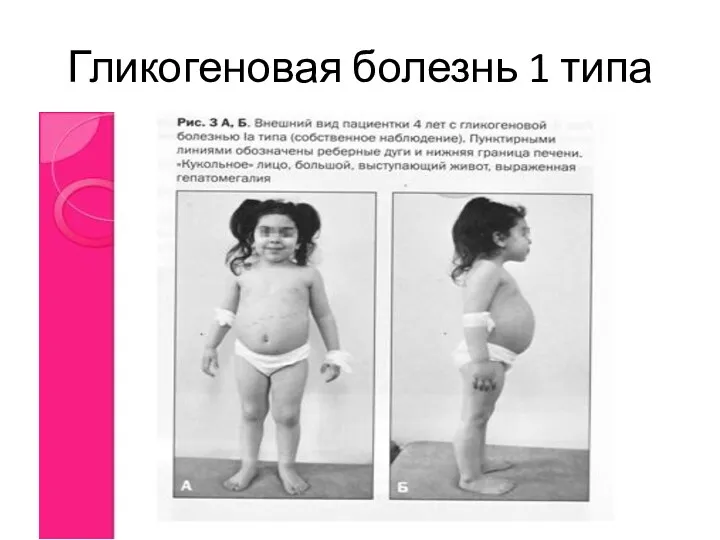
Гликогеновая болезнь 1 типа
Гликогеновая болезнь 1 типа

Гликогеноз II типа –болезнь Помпе
дефект лизосомной альфа-1,4- глюкозидазы
гликоген глюкоза
Симптомы проявляются
Гликогеноз II типа –болезнь Помпе
дефект лизосомной альфа-1,4- глюкозидазы
гликоген глюкоза
Симптомы проявляются

Гликогеноз III типа – болезнь Кори, болезнь Форбса -дефект амило-1,6- глюкозидазы
Страдает
Гликогеноз III типа – болезнь Кори, болезнь Форбса -дефект амило-1,6- глюкозидазы
Страдает

Гликогеноз V типа –болезнь Мак-Ардла
недостаточность миофосфорилазы
Гликоген + Н3РО4 Глюкозо-1 фосфат
Мышечные боли
Гликогеноз V типа –болезнь Мак-Ардла
недостаточность миофосфорилазы
Гликоген + Н3РО4 Глюкозо-1 фосфат
Мышечные боли
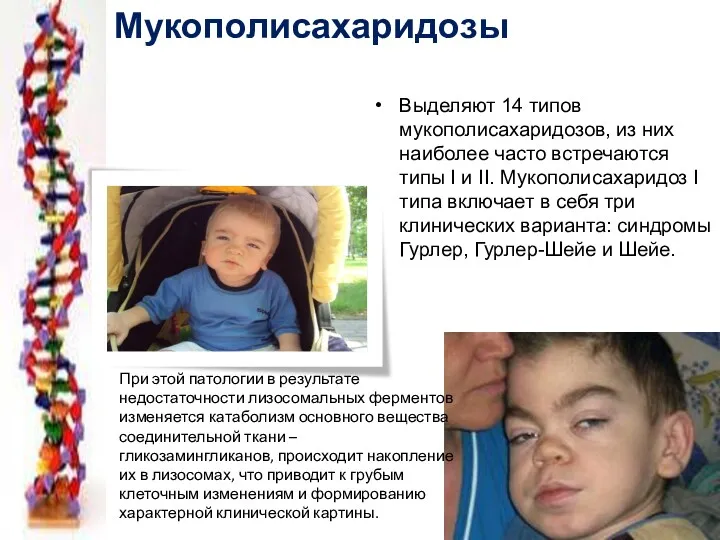
Выделяют 14 типов мукополисахаридозов, из них наиболее часто встречаются типы I
Выделяют 14 типов мукополисахаридозов, из них наиболее часто встречаются типы I

Болезнь
Гурлера Идуронидаза Дерматансульфаты
Гепарансульфаты Гепарансульфаты
Болезнь
Гюнтера Идуронатсульфатаза Дерматансульфаты
Болезнь
Моркио
Болезнь
Гурлера Идуронидаза Дерматансульфаты
Гепарансульфаты Гепарансульфаты
Болезнь
Гюнтера Идуронатсульфатаза Дерматансульфаты
Болезнь
Моркио
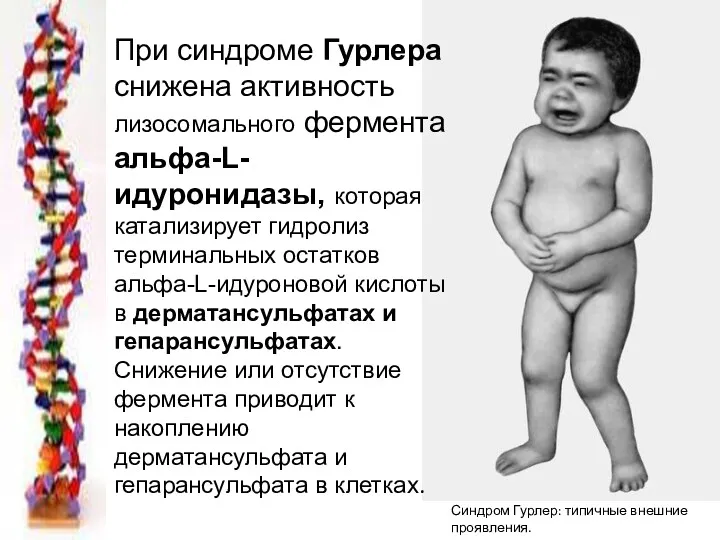
Синдром Гурлер: типичные внешние
проявления.
При синдроме Гурлера снижена активность лизосомального фермента
Синдром Гурлер: типичные внешние
проявления.
При синдроме Гурлера снижена активность лизосомального фермента
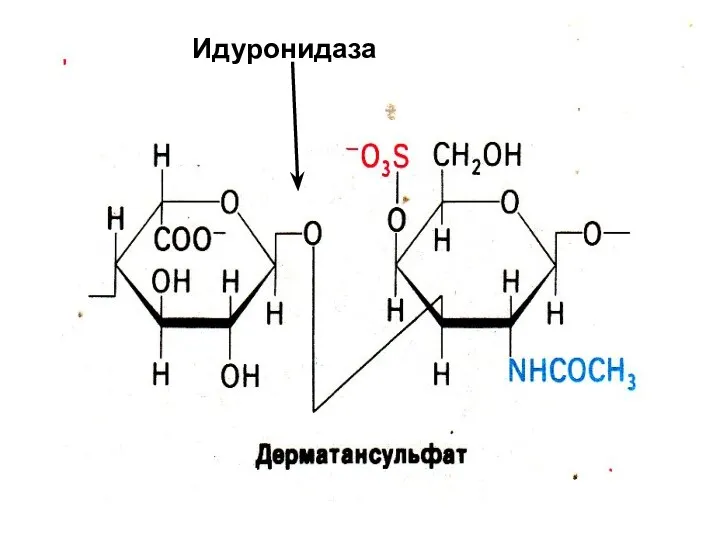
Идуронидаза
Идуронидаза

Синдром Гурлера-
дефект α-идуронидазы,
Грубая задержка
психомоторного
развития,
мегалоцефалия,
короткая шея,
запавшее переносье,
макроглоссия,
пупочная грыжа,
контрактуры
Синдром Гурлера-
дефект α-идуронидазы,
Грубая задержка
психомоторного
развития,
мегалоцефалия,
короткая шея,
запавшее переносье,
макроглоссия,
пупочная грыжа,
контрактуры
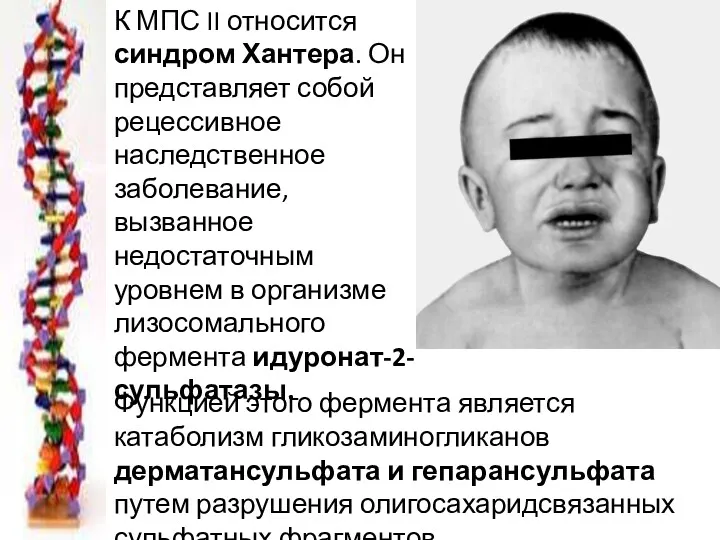
К МПС II относится синдром Хантера. Он представляет собой рецессивное наследственное
К МПС II относится синдром Хантера. Он представляет собой рецессивное наследственное
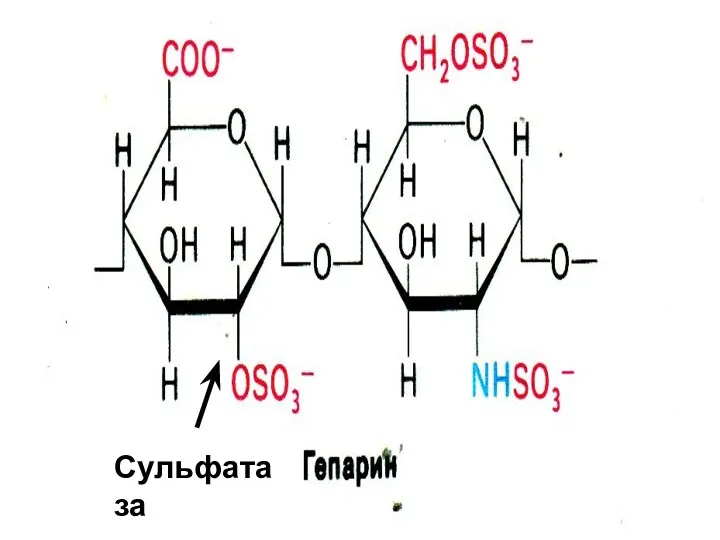
Сульфатаза
Сульфатаза

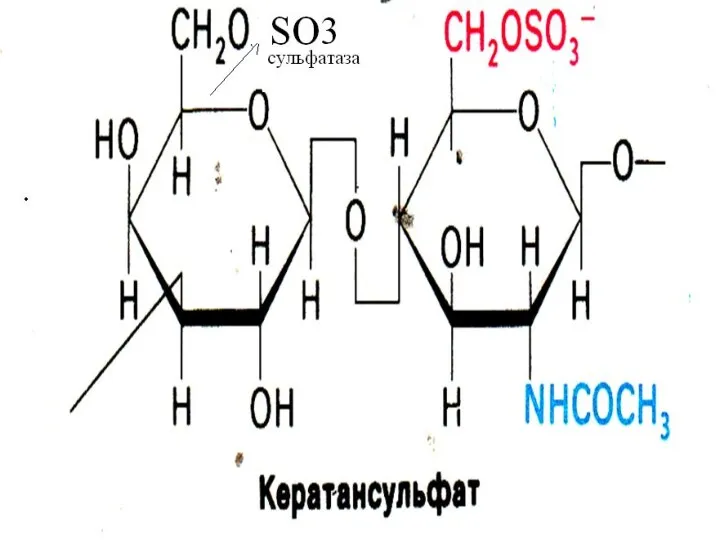
Синдром Моркио
Дефект галактозо-6-сульфатазы, нарушение распада кератансульфатов.
(Галактоза- N-ацетилглюкозамин-сульфат)
Симптомы: преимущественное поражение скелета,
Синдром Моркио
Дефект галактозо-6-сульфатазы, нарушение распада кератансульфатов.
(Галактоза- N-ацетилглюкозамин-сульфат)
Симптомы: преимущественное поражение скелета,

Терапия МПС
Ферментозаместительная терапия
Элапраза
Ларонидаза (альдуразим)
Трансплантация гемопоэтических стволовых клеток
Генотерапия
( с применением вирусных векторов)
Терапия МПС
Ферментозаместительная терапия
Элапраза
Ларонидаза (альдуразим)
Трансплантация гемопоэтических стволовых клеток
Генотерапия
( с применением вирусных векторов)

Ферментозамещающая терапия мукополисахаридозов
Ферментозамещающая терапия мукополисахаридозов

Энзимопатии липидного обмена
Энзимопатии липидного обмена

Энзимопатии липидного обмена
Сфинголипидозы
Болезнь Гоше (цереброзидоз)
недостаточность бета-гликозидазы
Церамид- олигосахарид- гексозамин
недостаточность гексаминидазы
Болезнь Тея –
Энзимопатии липидного обмена
Сфинголипидозы
Болезнь Гоше (цереброзидоз)
недостаточность бета-гликозидазы
Церамид- олигосахарид- гексозамин
недостаточность гексаминидазы
Болезнь Тея –
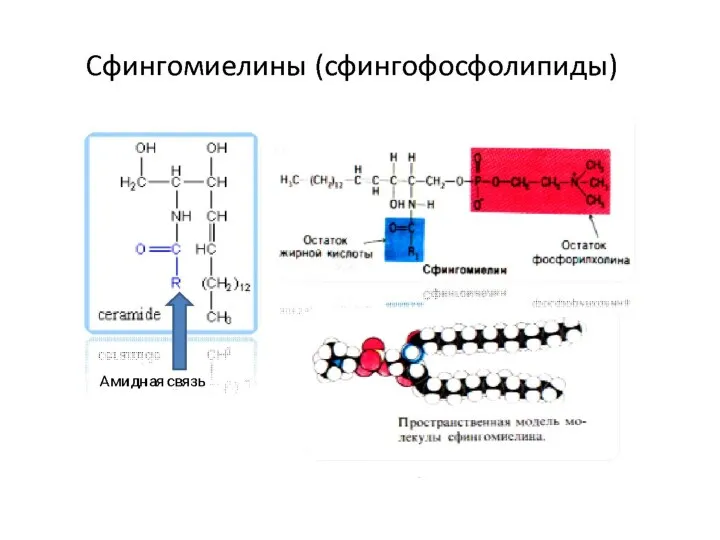
Структура сфингомиелинов
Структура сфингомиелинов
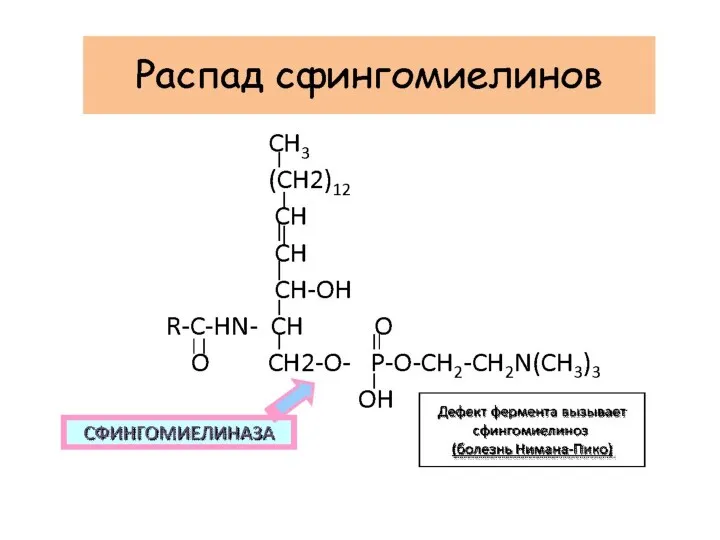

Сфингомиелиноз –болезнь Нимана-Пика
недостаточность лизосомной сфингомиелиназы
Церамид - фосфат- холин
Младенческий вариант А
Сфингомиелиноз –болезнь Нимана-Пика
недостаточность лизосомной сфингомиелиназы
Церамид - фосфат- холин
Младенческий вариант А

- манифестация основных симптомов заболевания на первом году жизни;
- грубые черты
- манифестация основных симптомов заболевания на первом году жизни;
- грубые черты
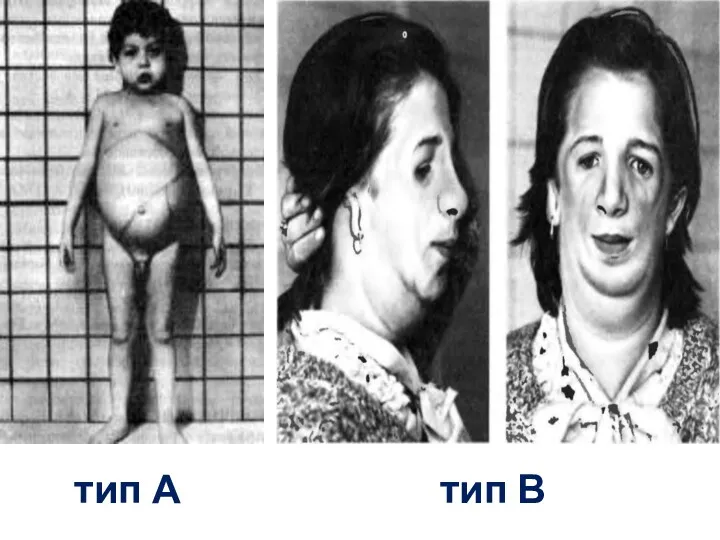
тип А
тип В
тип А
тип В
- манифестация основных симптомов заболевания на первом году жизни;
- грубые черты
- манифестация основных симптомов заболевания на первом году жизни;
- грубые черты
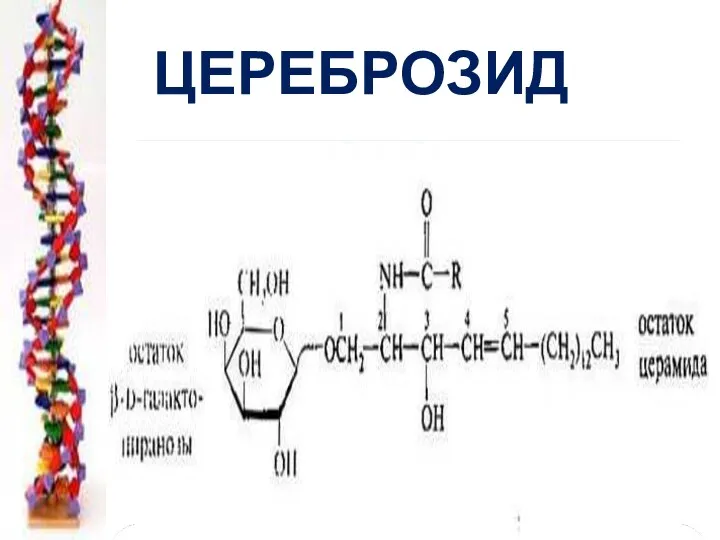
ЦЕРЕБРОЗИД
ЦЕРЕБРОЗИД
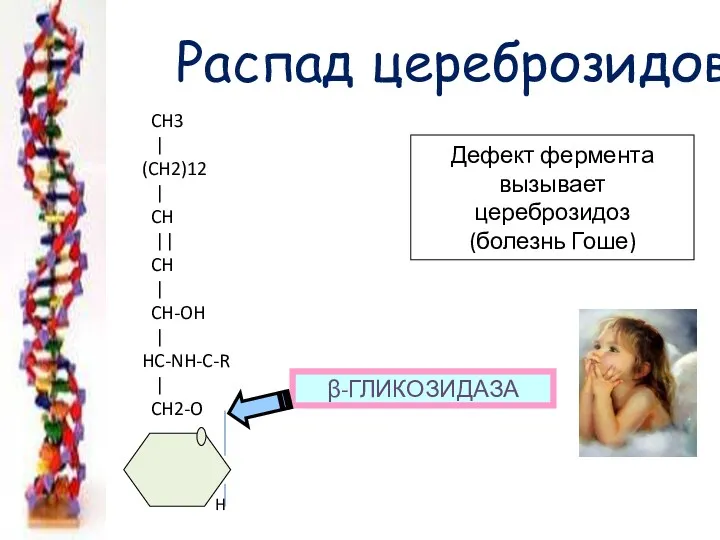
Распад цереброзидов
Дефект фермента
вызывает цереброзидоз
(болезнь Гоше)
β-ГЛИКОЗИДАЗА
CH3
|
(CH2)12
|
CH
||
Распад цереброзидов
Дефект фермента
вызывает цереброзидоз
(болезнь Гоше)
β-ГЛИКОЗИДАЗА
CH3
|
(CH2)12
|
CH
||

Цереброзидоз –болезнь Гоше
недостаточность лизосомальной β-гликозидазы
Церамид - галактоза
Накапливание цереброзида в ретикулоэндотелиальных
Цереброзидоз –болезнь Гоше
недостаточность лизосомальной β-гликозидазы
Церамид - галактоза
Накапливание цереброзида в ретикулоэндотелиальных

Ганглиозидоз -болезнь Тея-Сакса
недостаточность лизосомной гексаминидазы А и В
Церамид - олигосахарид
Ганглиозидоз -болезнь Тея-Сакса
недостаточность лизосомной гексаминидазы А и В
Церамид - олигосахарид
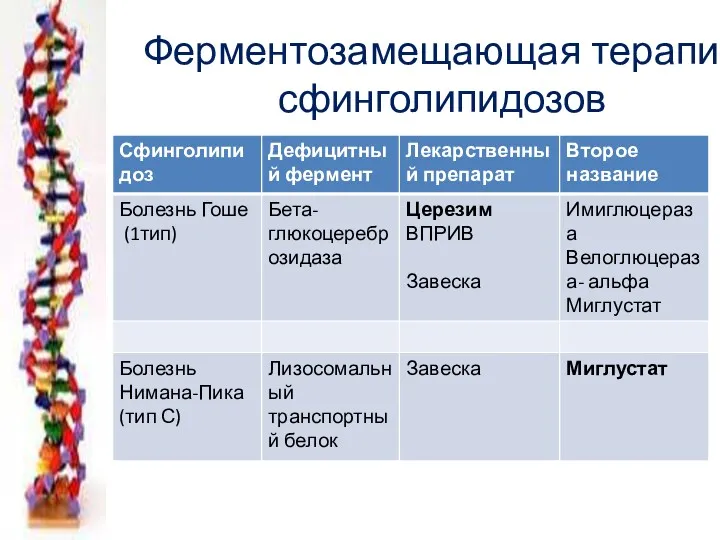
Ферментозамещающая терапия сфинголипидозов
Ферментозамещающая терапия сфинголипидозов

Аферментозы обмена нуклеотидов
болезнь Леш-Нихана
Недостаточность гипоксантин-гуанин-фосфорибозилтрансферазы
Гуанин + ФРПФ ГМФ
мочевая кислота
Оротатацидурия –
Аферментозы обмена нуклеотидов
болезнь Леш-Нихана
Недостаточность гипоксантин-гуанин-фосфорибозилтрансферазы
Гуанин + ФРПФ ГМФ
мочевая кислота
Оротатацидурия –

Энзимопатии обмена гемоглобина
Фавизм (гемолитическая анемия)
недостаточность глюкозо-6-фосфатдегидрогеназы
Врождённая метгемоглобинопатия –
недостаточность НАДН-цитохром В5-редуктазы
Порфирии –
недостаточность
Энзимопатии обмена гемоглобина
Фавизм (гемолитическая анемия)
недостаточность глюкозо-6-фосфатдегидрогеназы
Врождённая метгемоглобинопатия –
недостаточность НАДН-цитохром В5-редуктазы
Порфирии –
недостаточность

Порфирии-
нарушение активности ферментов синтеза гема
Клинически выделяют :
печёночные порфирии с выраженными неврологическими
Порфирии-
нарушение активности ферментов синтеза гема
Клинически выделяют :
печёночные порфирии с выраженными неврологическими

Классификация порфирий
Классификация порфирий

Нарушения обмена билирубина
Синдром Криглера – Найяра
(неконьюгированная желтуха)
Отсутствие глюкуронилтрансферазы
Гипербилирубинемия за
Нарушения обмена билирубина
Синдром Криглера – Найяра
(неконьюгированная желтуха)
Отсутствие глюкуронилтрансферазы
Гипербилирубинемия за

Транзиторная желтуха новорожденных
Усилен гемолиз эритроцитов
Снижено содержание альбуминов
Снижен захват билирубина печенью
Снижена активность
Транзиторная желтуха новорожденных
Усилен гемолиз эритроцитов
Снижено содержание альбуминов
Снижен захват билирубина печенью
Снижена активность

Митохондриальные болезни
Проявляются нарушением тканевого дыхания, миопатией, поражением нервной системы, печени, сердца,
Митохондриальные болезни
Проявляются нарушением тканевого дыхания, миопатией, поражением нервной системы, печени, сердца,

Нарушения обмена жирных кислот
Группа тяжёлых наследственных болезней, характеризующихся высокой смертностью, преимущественным
Нарушения обмена жирных кислот
Группа тяжёлых наследственных болезней, характеризующихся высокой смертностью, преимущественным

Клинические симптомы
нарушения обмена жирных кислот
Неонатальная
Детская
Поздняя формы
Энцефалопатия (вялость, сонливость, кома), рвота, кардиомиопатия,
Клинические симптомы
нарушения обмена жирных кислот
Неонатальная
Детская
Поздняя формы
Энцефалопатия (вялость, сонливость, кома), рвота, кардиомиопатия,

Биохимические сдвиги
при нарушении обмена жирных кислот
Не происходит бета-окисления, активируется омега-окисление
Биохимические сдвиги
при нарушении обмена жирных кислот
Не происходит бета-окисления, активируется омега-окисление

Диагностика
нарушений обмена жирных кислот
Определение уровня карнитина
Определение экскреции органических кислот
Снижение активности
Диагностика
нарушений обмена жирных кислот
Определение уровня карнитина
Определение экскреции органических кислот
Снижение активности

Лечение
нарушений обмена жирных кислот
Диетотерапия (исключение голода, обогащение рациона углеводами при снижении
Лечение
нарушений обмена жирных кислот
Диетотерапия (исключение голода, обогащение рациона углеводами при снижении
 Возбудители хронических бактериальных инфекций с поражением слизистой рта и губ ( туберкулез, лепра)
Возбудители хронических бактериальных инфекций с поражением слизистой рта и губ ( туберкулез, лепра) Противоэпидемические мероприятия в очагах особо-опасных инфекций
Противоэпидемические мероприятия в очагах особо-опасных инфекций Заболевания век, слезных органов и орбиты. Конъюнктивит, трахома
Заболевания век, слезных органов и орбиты. Конъюнктивит, трахома Неотложные состояния в психиатрии и наркологии (часть 1)
Неотложные состояния в психиатрии и наркологии (часть 1) Гигиенические требования к водоснабжению дошкольных образовательных организаций
Гигиенические требования к водоснабжению дошкольных образовательных организаций Гигиена кожи
Гигиена кожи Хронический гастрит у детей
Хронический гастрит у детей Противоестественный задний проход и колостома
Противоестественный задний проход и колостома Гостра ревматична лихоманка у дітей
Гостра ревматична лихоманка у дітей Витамины и биостимуляторы
Витамины и биостимуляторы Общая пропедевтика мочевыделительной системы
Общая пропедевтика мочевыделительной системы Прикорм. Види і правила його введення змішане та штучне вигодовування
Прикорм. Види і правила його введення змішане та штучне вигодовування Ас қорыту жуйесіне әсер ететін дәрілер
Ас қорыту жуйесіне әсер ететін дәрілер Современные подходы к лечению аневризмы брюшной аорты
Современные подходы к лечению аневризмы брюшной аорты Аутоиммунный гепатит. Перекрест с первичным биллиарным холангитом
Аутоиммунный гепатит. Перекрест с первичным биллиарным холангитом Инфекция - ассоциированный (постинфекционный) гломерулонефрит
Инфекция - ассоциированный (постинфекционный) гломерулонефрит Правильная осанка. Нарушение осанки
Правильная осанка. Нарушение осанки Лейкоплакия. Этиологиясы, клиникасы, диагностикасы, емі
Лейкоплакия. Этиологиясы, клиникасы, диагностикасы, емі Ретинопатия у новорожденных
Ретинопатия у новорожденных Отруєння важкими металами
Отруєння важкими металами Терминальды жағдайлардың пайда болу себебіне байланысты өкпе-жүрек реанимациясын орындау ерекшелігі
Терминальды жағдайлардың пайда болу себебіне байланысты өкпе-жүрек реанимациясын орындау ерекшелігі Особенности вскармливания детей раннего возраста
Особенности вскармливания детей раннего возраста Полное обследование новорожденного. (Модуль 1)
Полное обследование новорожденного. (Модуль 1) Анатомия коронарных артерий
Анатомия коронарных артерий Лечебная физическая культура
Лечебная физическая культура Адамның тұқымқуалайтын патологиясындағы тұқымқуалаушылық пен ортаның рөлі
Адамның тұқымқуалайтын патологиясындағы тұқымқуалаушылық пен ортаның рөлі Фетальный алкогольный синдром
Фетальный алкогольный синдром Вирусные гепатиты B, C, D
Вирусные гепатиты B, C, D