Причины, клинические проявления проблем пациентов детского возраста при наследственных и врожденных заболеваниях презентация
- Причины, клинические проявления проблем пациентов детского возраста при наследственных и врожденных заболеваниях

Содержание
- 2. Наследственные болезни - заболевания, обусловленные хромосомными и генными мутациями. Их более 6000 Врожденные болезни - заболевания,
- 3. При возникновении мутации в клетке на ранних стадиях онтогенеза, из неё будут развиваться ткани, все клетки
- 4. Генеративные мутации Моногенные - мутации в одном гене Общая частота генных болезней в популяции составляет 1-2%
- 5. НАСЛЕДСТВЕННАЯ ПАТОЛОГИЯ ГЕННЫЕ БОЛЕЗНИ
- 7. КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ Генные болезни; Хромосомные болезни; Болезни с наследственной предрасположенностью (мультифакториальные болезни); Группа генетических болезней,
- 8. ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ Ранняя манифестация; Хроническое прогредиентное течение; Относительная резистентность к терапии; Множественность поражения;
- 9. ГЕННЫЕ БОЛЕЗНИ это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на генном уровне. Число известных
- 10. ПРИМЕРЫ РАСПРОСТРАНЕННЫХ ГЕННЫХ БОЛЕЗНЕЙ Нейрофиброматоз Синдром Марфана Болезнь Олбрайта Дизостозы Отосклероз Пароксизмальная миоплегия Талассемия Семейная гипехолестеринемия
- 14. СИНДРОМ МАРФАНА Это одна из наследственных форм врожденной генерализованной патологии соединительной ткани, впервые описана в 1886
- 15. - наследственное заболевание соединительной ткани, проявляющееся изменениями скелета: высоким ростом с относительно коротким туловищем, длинными паукообразными
- 16. Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечно-сосудистых осложнений, но и появлению
- 17. СИНДРОМ МАРФАНА Симптом «большого пальца» Арахнодактелия (длинные пальцы) Симптом «запястья» «Сандалевидная щель»
- 19. Арахнодактилия – удлинение суставов
- 21. Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от аневризма аорты. Единственная компенсация –
- 22. ИЗВЕСТНЫЕ ЛЮДИ С СИНДРОМОМ МАРФАНА Эхнатон Н. Паганини Ш. де Голль А. Линкольн
- 23. ФЕНИЛКЕТОНУРИЯ (ФЕНИЛПИРОВИНОГРАДНАЯ ОЛИГОФРЕНИЯ) Фенилкетонурия (ФКУ) – это она из самых частых форм наследственных дефектов обмена аминокислот.
- 24. Классическая форма фенилкетонурии, ребенок в возрасте 1 год 7 мес. Грубая задержка психомоторного развития, судороги (до
- 25. «обречённая на вегетарианство»
- 27. фенотипически проявляется повышенной ломкостью костей, вследствие нарушения остеогенеза, изменениями в суставах, глухотой, голубыми склерами, аномалиями зубов.
- 29. врожденное поражение скелета врождённая болезнь, характеризующаяся нарушением развития хрящевой ткани; проявляется карликовостью, короткими конечностями при обычной
- 30. Человек, закончивших свой рост, достигает 30 - 41 см
- 31. Причины мутации в настоящее время не известны
- 33. СИНДРОМ ХОЛТ – ОРАМА (СИНДРОМ РУКА – СЕРДЦЕ) Синдром Холт – Орама представляет собой моногенный синдром
- 34. МУКОВИСЦИДОЗ - наследственное заболевание желез внутренней секреции, а также поджелудочной железы и печени, характеризующееся, в первую
- 35. Новорожденный с муковисцидозом
- 36. Муковисцидоз - это системное наследственное заболевание , обычно проявляющееся в детстве, хотя в 4% случаев диагноз
- 37. ГОМОЦИСТИНУРИЯ – НАРУШЕНИЕ МЕТАБОЛИЗМА МЕТИОНИНА При гомоцистинурии, как и при других наследственных нарушениях обмена веществ, в
- 38. Ира Д., 7 лет. Диагноз: гомоцистинурия
- 39. Подвывих хрусталика при гомоцистинурии
- 40. СИНДРОМ ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ Относится к редким заболеваниям, для которого характерен своеобразный симтомокомплекс – сочетание пигментного ретинита, ожирение,
- 41. Дети с синдромом ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ полидактилия
- 42. Наследственные болезни обмена веществ соединительной ткани. МУКОПОЛИСАХАРИДОЗЫ Под термином «мукополисахаридозы» объединяется ряд патологических процессов, в основе
- 43. МУКОПОЛИСАХАРИДОЗ I ТИПА (СИНДРОМ ГУРЛЕР) Заболевание характеризуется грубыми поражениями опорно-двигательного аппарата, выраженной умственной отсталостью, помутнением роговицы
- 44. МУКОПОЛИСАХАРИДОЗ II ТИПА (СИНДРОМ ГУНТЕРА) В клинической картине на первый план выступает костные деформации и тугоподвижность
- 45. Изменения костной ткани при мукополисахаридозах
- 46. МУКОПОЛИСАХАРИДОЗ V ТИПА (СИНДРОМ ШЕЙЕ) Клиническая картина заболевания складывается из наличия характерных грубых внешних черт, тугоподвижности
- 47. ПРОГЕРИЯ (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений кожи, внутренних органов, обусловленных преждевременным
- 48. Описана в 1886 г. Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения в 8-10 раз. Дети
- 51. ИХТИОЗ (греч. - рыба) — наследственный дерматоз, характеризующийся диффузным нарушением ороговения по типу гиперкератоза, проявляется образованием
- 56. Гиперпигментирванные пятна выявляются с рождения на различных участках кожи, варьируя по размеру и окраске, часто кофе
- 57. Чаще наблюдается нейрофиброматоз I типа ( болезнь Реклингхаузена ) - аутосомно-доминантное заболевание, характеризующееся наличием множественных пигментных
- 58. НЕЙРОФИБРОМАТОЗ У больного этим заболеванием наблюдается: слоновость левой верхней конечности, обусловленная множественными нейрофиброматозными узлами.
- 59. Почти у всех больных на радужке обнаруживаются небольшие гематомы . Нейрофибромы могут подвергаться злокачественной трансформации с
- 60. НЕЙРОФИРОМАТОЗ Множественные нейрофибромы
- 64. АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ (ВРАЖДЕННАЯ ГИПЕРПЛАЗИЯ КОРЫ НАДПОЧЕЧНИКОВ) Адреногенитальный синдром (АГС) относится к группе наследственных нарушений биосинтеза стероидных
- 65. Новорожденная девочка с двойственным строением наружных половых органов Мальчики шести лет (справа – опережение полового и
- 68. ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МЫШЕЧНАЯ ДИСТРОФИЯ (ДЮШЕННА) Это одна из самых частых форм наследственных нервно – мышечных заболеваний. Впервые
- 69. А,Б – Ряд последовательных движений при принятии вертикального положения симптом «лестницы» А Б В В –
- 72. Странное племя людей-страусов (сапади) в Центральной Африке отличает от прочих обитателей Земли удивительное свойство: на ногах
- 74. Синдром Ангельмана СИНДРОМ АНГЕЛЬМАНА (СА) – это нейро-генетическое заболевание, характеризующееся интеллектуальной и физической задержкой развития, нарушениями
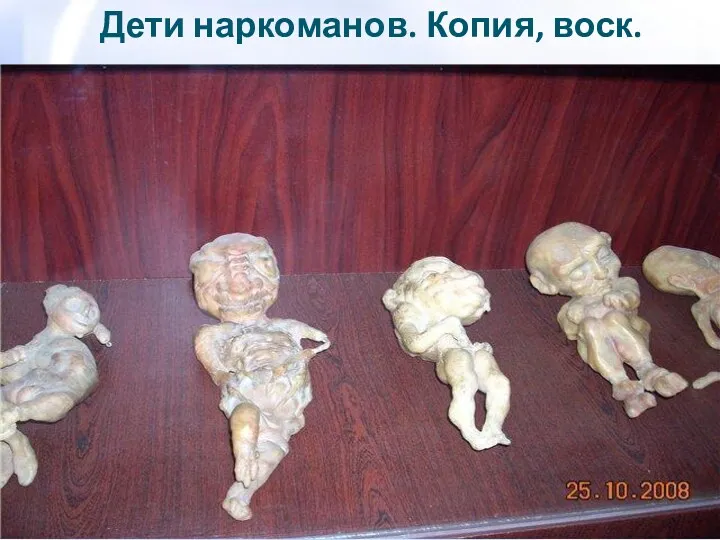
- 78. Дети наркоманов. Копия, воск.
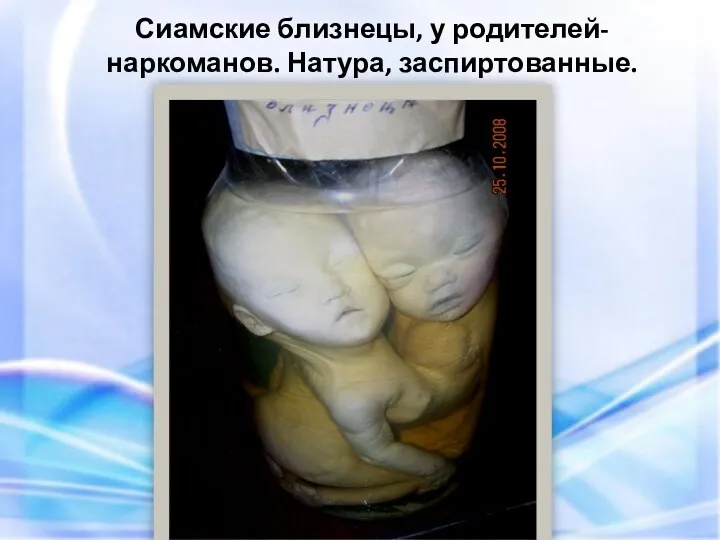
- 79. Сиамские близнецы, у родителей-наркоманов. Натура, заспиртованные.
- 80. Дети у родителей больных наследственными заболеваниями. Копия, воск.
- 81. Ребенок, родившийся в результате инцеста(кровосмешения родственников). Натура, заспиртован. Ответьте на проблемный вопрос. Почему в близкородственных браках
- 82. Ребенок, родившийся в семье чернобыльцев. Натура, мумия.
- 83. Человек-циклоп, и женщина-слон. Жили в 19 веке. Копия, воск.
- 84. Согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом, можно говорить о многообразии видов
- 85. Если принять, что у человека примерно 100 000 генов и каждый ген может мутировать и контролировать
- 86. Вся история развития человека есть непрерывная цепь мутаций в нем. Мутации происходят постоянно как отрицательные, так
- 88. Скачать презентацию
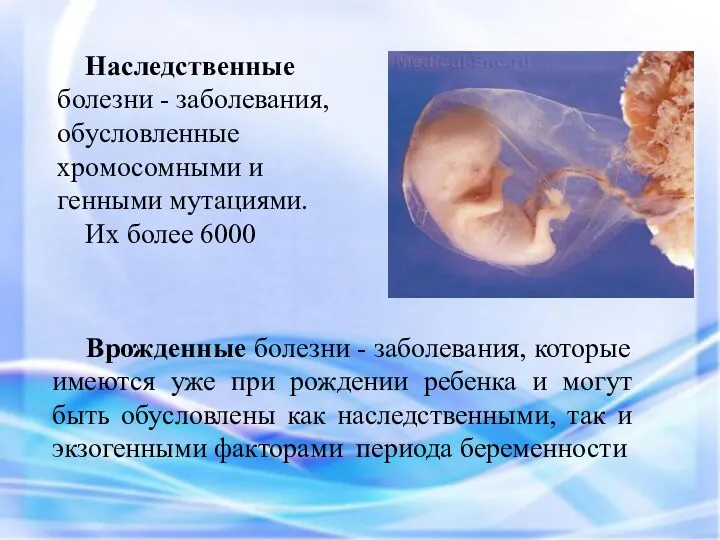
Наследственные болезни - заболевания, обусловленные хромосомными и генными мутациями.
Их более 6000
Наследственные болезни - заболевания, обусловленные хромосомными и генными мутациями.
Их более 6000
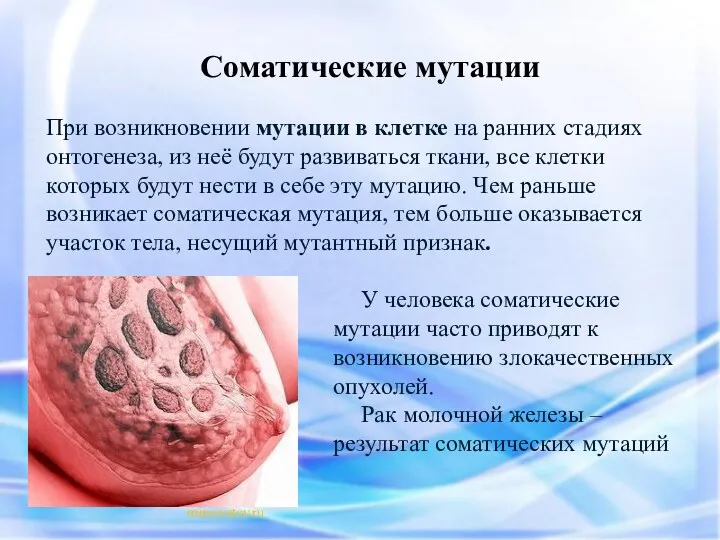
При возникновении мутации в клетке на ранних стадиях онтогенеза, из неё
При возникновении мутации в клетке на ранних стадиях онтогенеза, из неё

Генеративные мутации
Моногенные - мутации в одном гене
Общая частота генных болезней в
Генеративные мутации
Моногенные - мутации в одном гене
Общая частота генных болезней в

НАСЛЕДСТВЕННАЯ ПАТОЛОГИЯ
ГЕННЫЕ БОЛЕЗНИ
НАСЛЕДСТВЕННАЯ ПАТОЛОГИЯ
ГЕННЫЕ БОЛЕЗНИ

КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Генные болезни;
Хромосомные болезни;
Болезни с наследственной предрасположенностью (мультифакториальные болезни);
Группа
КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Генные болезни;
Хромосомные болезни;
Болезни с наследственной предрасположенностью (мультифакториальные болезни);
Группа

ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Ранняя манифестация;
Хроническое прогредиентное течение;
Относительная резистентность к терапии;
Множественность
ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Ранняя манифестация;
Хроническое прогредиентное течение;
Относительная резистентность к терапии;
Множественность

ГЕННЫЕ БОЛЕЗНИ
это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями
ГЕННЫЕ БОЛЕЗНИ
это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями

ПРИМЕРЫ РАСПРОСТРАНЕННЫХ ГЕННЫХ БОЛЕЗНЕЙ
Нейрофиброматоз
Синдром Марфана
Болезнь Олбрайта
Дизостозы
Отосклероз
Пароксизмальная миоплегия
Талассемия
Семейная гипехолестеринемия
Несовершенный остеогенез
Болезнь
ПРИМЕРЫ РАСПРОСТРАНЕННЫХ ГЕННЫХ БОЛЕЗНЕЙ
Нейрофиброматоз
Синдром Марфана
Болезнь Олбрайта
Дизостозы
Отосклероз
Пароксизмальная миоплегия
Талассемия
Семейная гипехолестеринемия
Несовершенный остеогенез
Болезнь
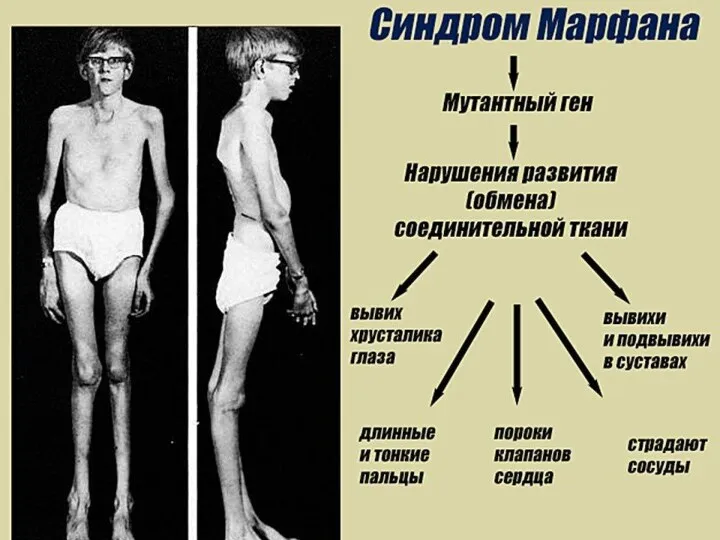
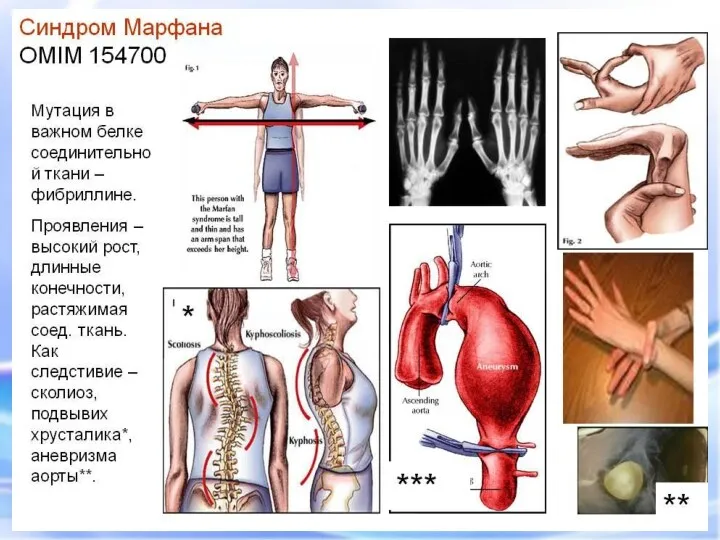
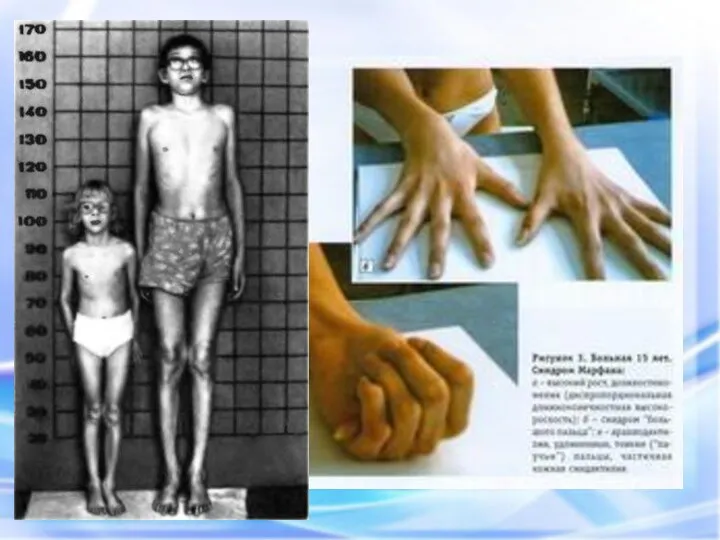
СИНДРОМ МАРФАНА
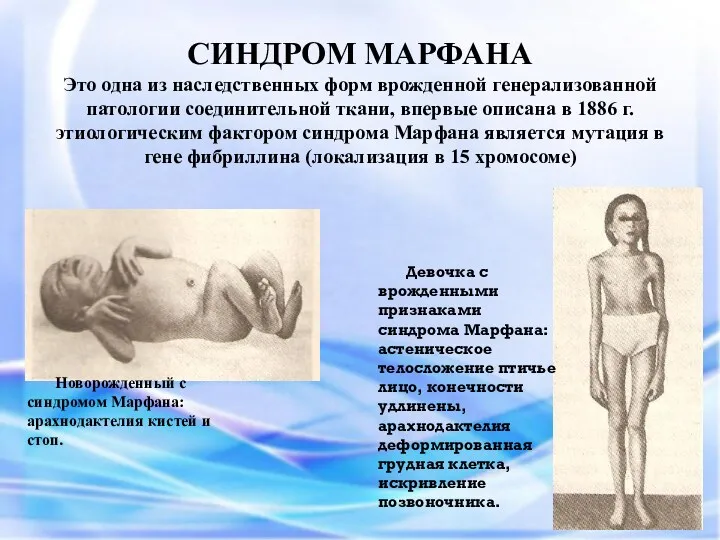
Это одна из наследственных форм врожденной генерализованной патологии соединительной ткани,
СИНДРОМ МАРФАНА Это одна из наследственных форм врожденной генерализованной патологии соединительной ткани,
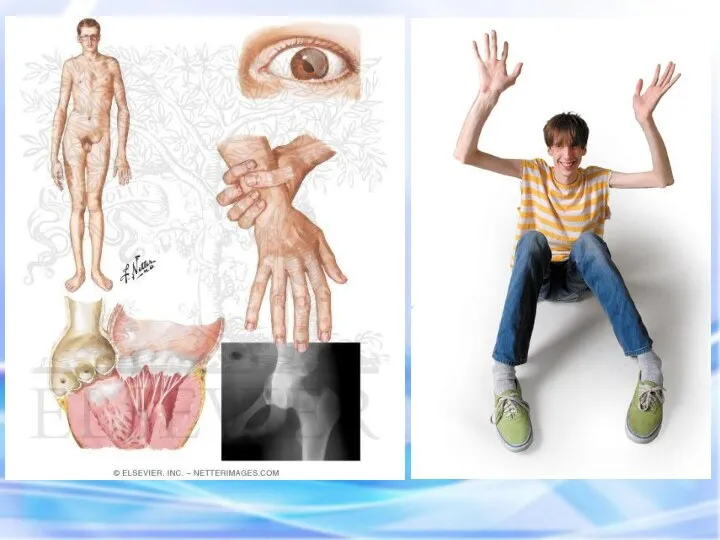
- наследственное заболевание соединительной ткани, проявляющееся изменениями скелета: высоким ростом
- наследственное заболевание соединительной ткани, проявляющееся изменениями скелета: высоким ростом
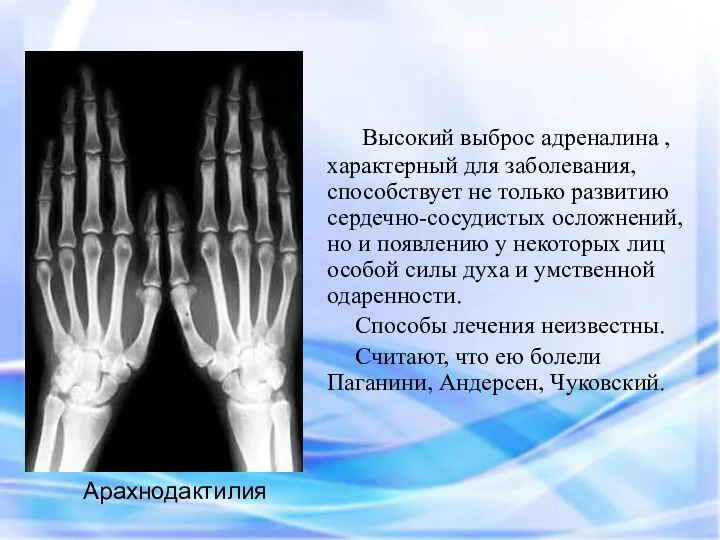
Высокий выброс адреналина , характерный для заболевания, способствует не только
Высокий выброс адреналина , характерный для заболевания, способствует не только
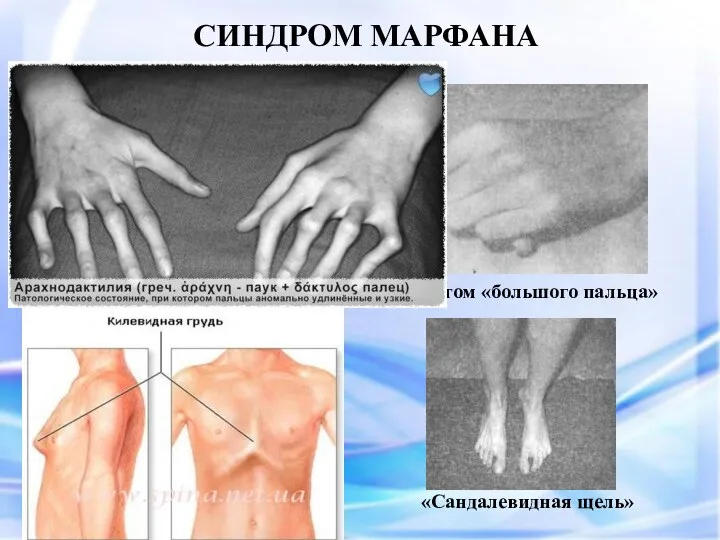
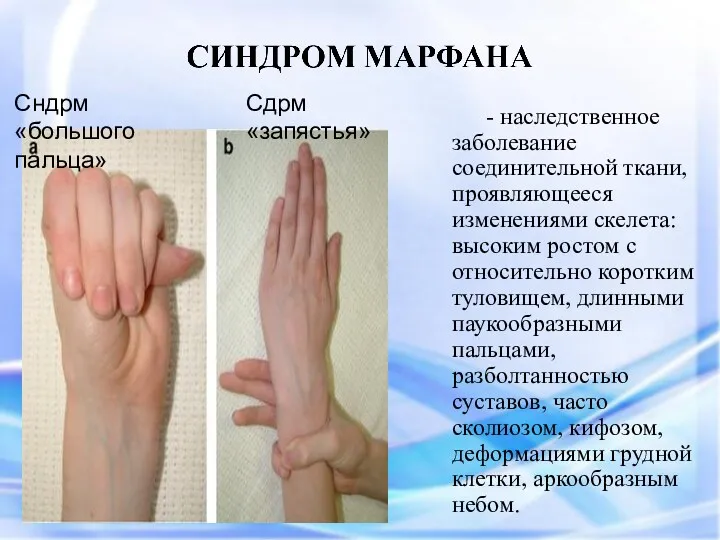
СИНДРОМ МАРФАНА
Симптом «большого пальца»
Арахнодактелия (длинные пальцы)
Симптом «запястья»
«Сандалевидная щель»
СИНДРОМ МАРФАНА
Симптом «большого пальца»
Арахнодактелия (длинные пальцы)
Симптом «запястья»
«Сандалевидная щель»
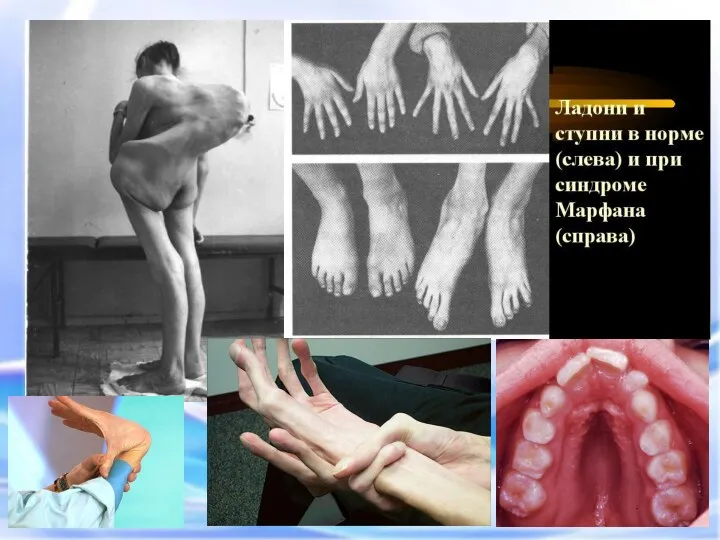
Арахнодактилия – удлинение суставов
Арахнодактилия – удлинение суставов
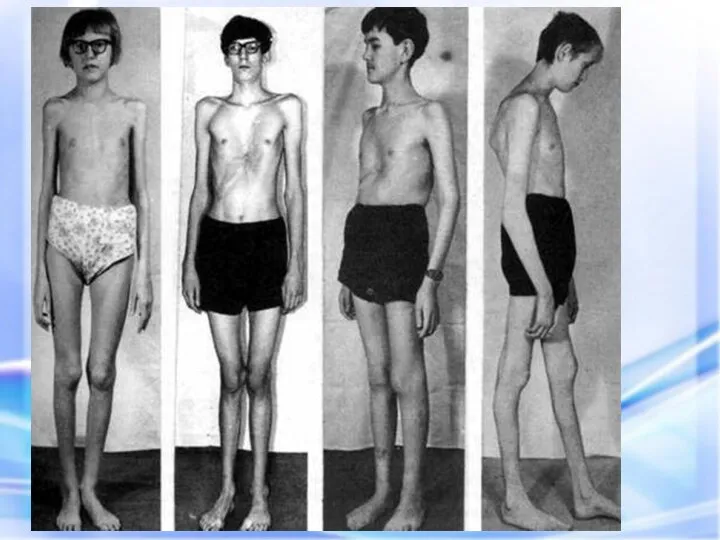

Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от
Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от

ИЗВЕСТНЫЕ ЛЮДИ С СИНДРОМОМ МАРФАНА
Эхнатон Н. Паганини
Ш. де Голль
ИЗВЕСТНЫЕ ЛЮДИ С СИНДРОМОМ МАРФАНА
Эхнатон Н. Паганини
Ш. де Голль
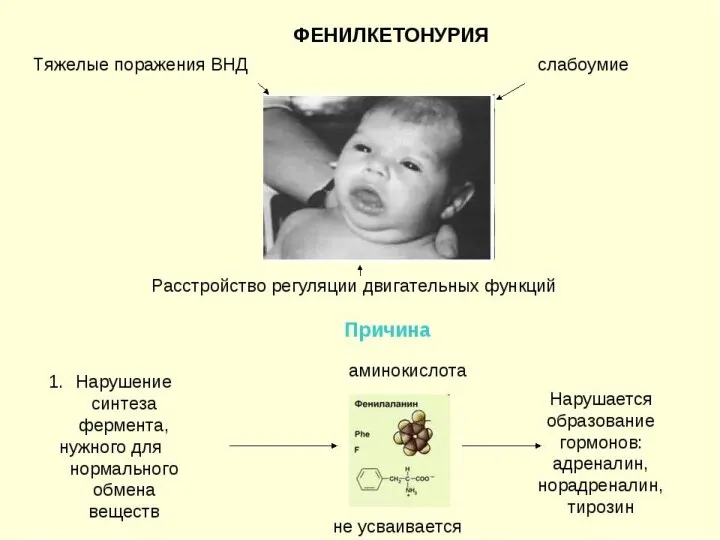
ФЕНИЛКЕТОНУРИЯ
(ФЕНИЛПИРОВИНОГРАДНАЯ ОЛИГОФРЕНИЯ)
Фенилкетонурия (ФКУ) – это она из самых частых форм
ФЕНИЛКЕТОНУРИЯ
(ФЕНИЛПИРОВИНОГРАДНАЯ ОЛИГОФРЕНИЯ)
Фенилкетонурия (ФКУ) – это она из самых частых форм

Классическая форма фенилкетонурии, ребенок в возрасте 1 год 7 мес.
Грубая задержка
Классическая форма фенилкетонурии, ребенок в возрасте 1 год 7 мес.
Грубая задержка

«обречённая на вегетарианство»
«обречённая на вегетарианство»

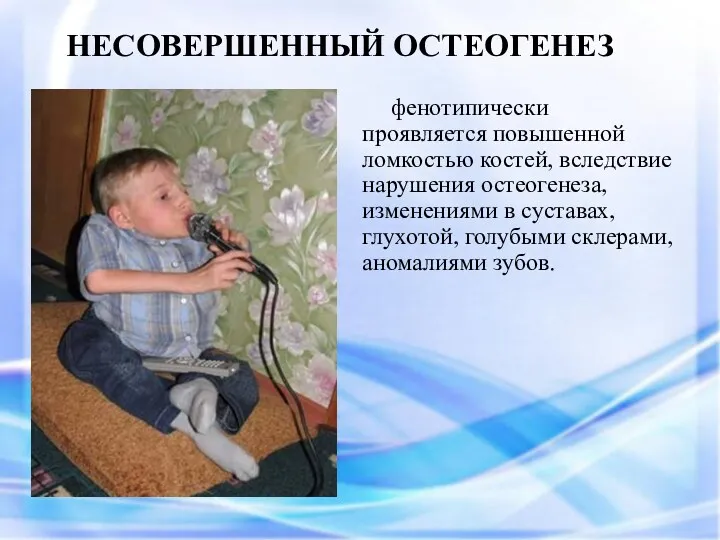
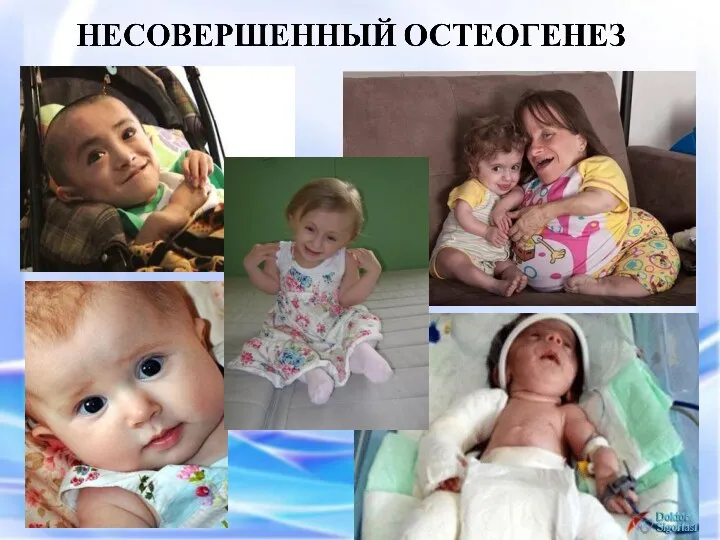
фенотипически проявляется повышенной ломкостью костей, вследствие нарушения остеогенеза, изменениями в суставах,
фенотипически проявляется повышенной ломкостью костей, вследствие нарушения остеогенеза, изменениями в суставах,

врожденное поражение скелета
врождённая болезнь, характеризующаяся нарушением развития хрящевой ткани; проявляется
врожденное поражение скелета
врождённая болезнь, характеризующаяся нарушением развития хрящевой ткани; проявляется
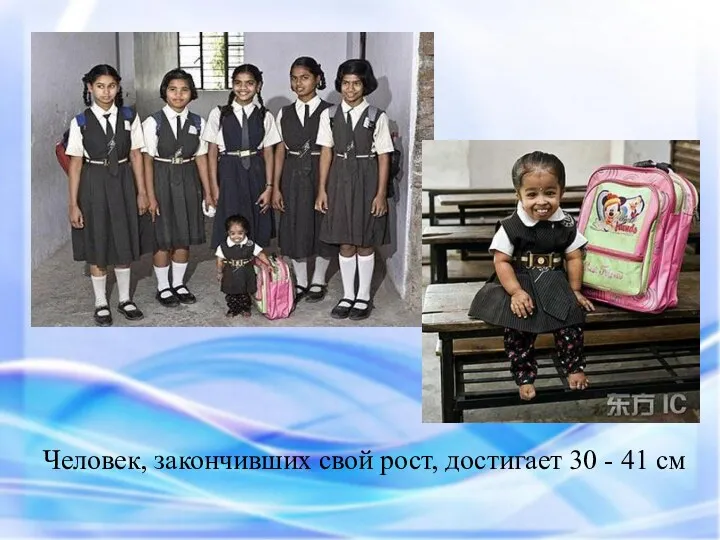
Человек, закончивших свой рост, достигает 30 - 41 см
Человек, закончивших свой рост, достигает 30 - 41 см

Причины мутации в настоящее время не известны
Причины мутации в настоящее время не известны
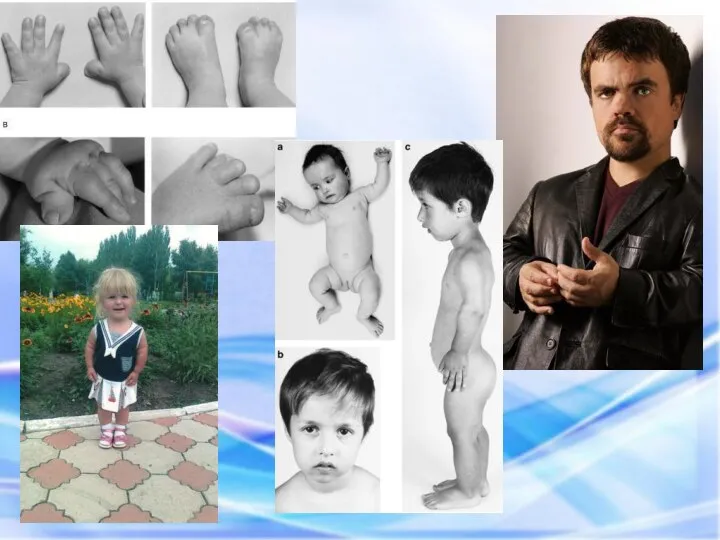
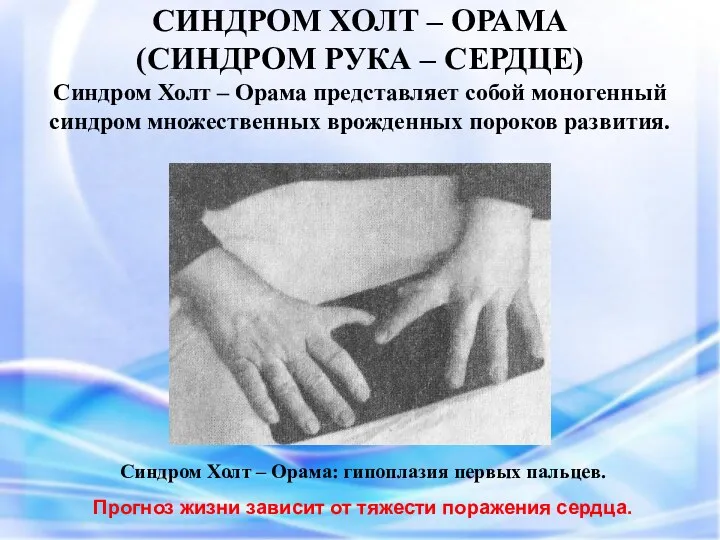
СИНДРОМ ХОЛТ – ОРАМА
(СИНДРОМ РУКА – СЕРДЦЕ)
Синдром Холт – Орама
СИНДРОМ ХОЛТ – ОРАМА (СИНДРОМ РУКА – СЕРДЦЕ) Синдром Холт – Орама
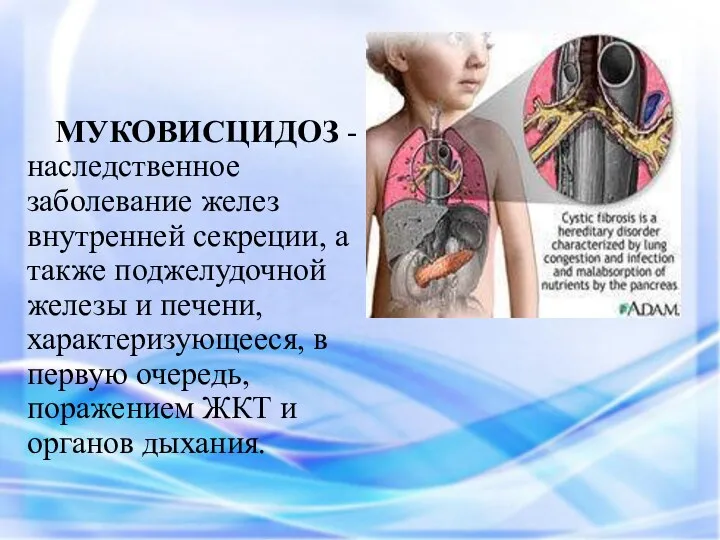
МУКОВИСЦИДОЗ - наследственное заболевание желез внутренней секреции, а также поджелудочной железы
МУКОВИСЦИДОЗ - наследственное заболевание желез внутренней секреции, а также поджелудочной железы
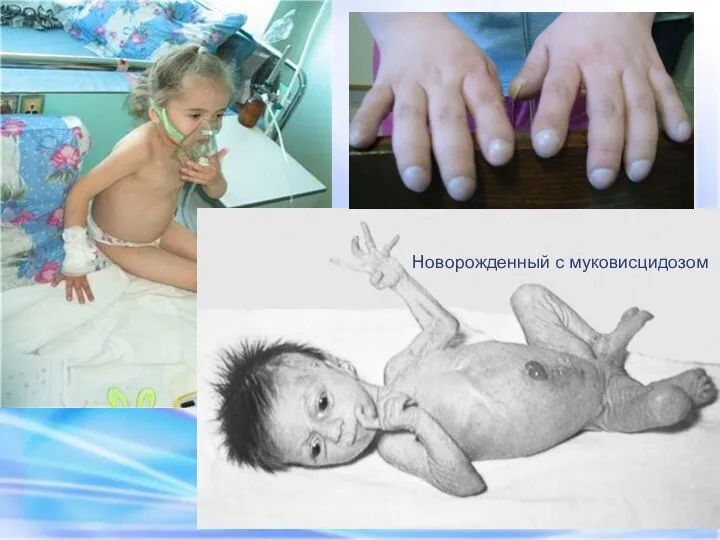
Новорожденный с муковисцидозом
Новорожденный с муковисцидозом
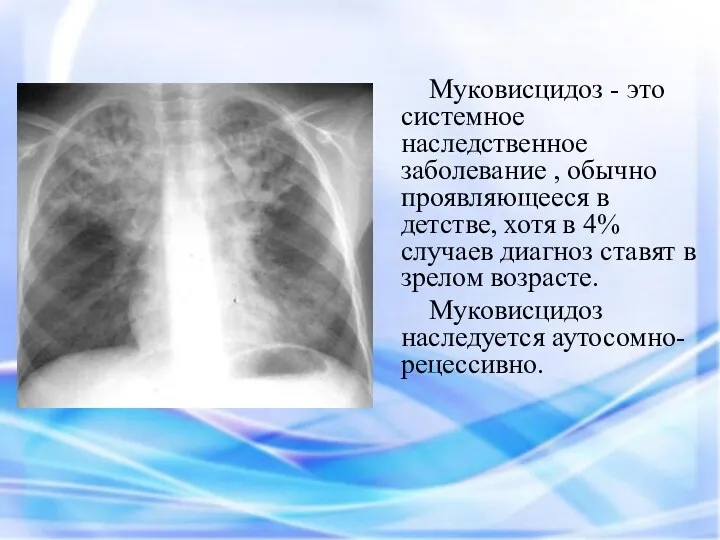
Муковисцидоз - это системное наследственное заболевание , обычно проявляющееся в детстве,
Муковисцидоз - это системное наследственное заболевание , обычно проявляющееся в детстве,

ГОМОЦИСТИНУРИЯ – НАРУШЕНИЕ МЕТАБОЛИЗМА МЕТИОНИНА
При гомоцистинурии, как и при других наследственных
ГОМОЦИСТИНУРИЯ – НАРУШЕНИЕ МЕТАБОЛИЗМА МЕТИОНИНА
При гомоцистинурии, как и при других наследственных
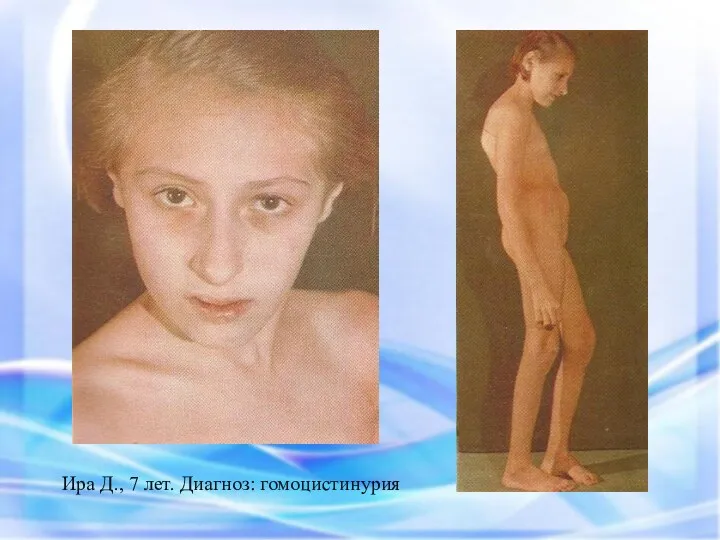
Ира Д., 7 лет. Диагноз: гомоцистинурия
Ира Д., 7 лет. Диагноз: гомоцистинурия

Подвывих хрусталика при гомоцистинурии
Подвывих хрусталика при гомоцистинурии

СИНДРОМ ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ
Относится к редким заболеваниям, для которого характерен своеобразный симтомокомплекс –
СИНДРОМ ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ
Относится к редким заболеваниям, для которого характерен своеобразный симтомокомплекс –
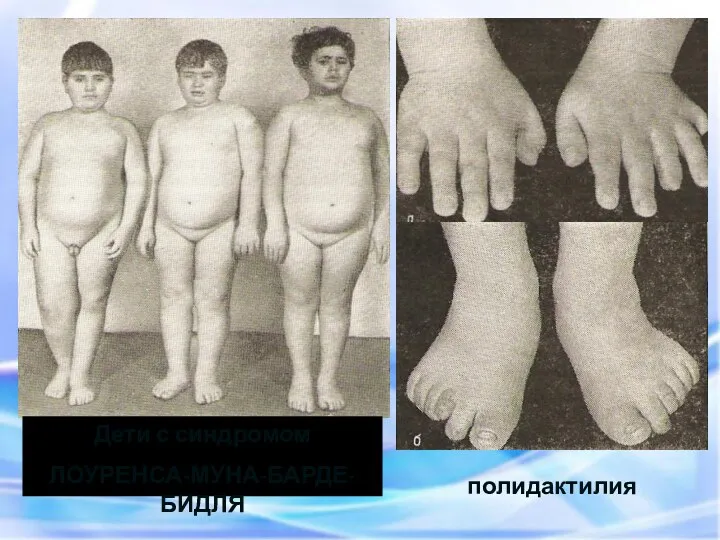
Дети с синдромом
ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ
полидактилия
Дети с синдромом
ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ
полидактилия

Наследственные болезни обмена веществ соединительной ткани.
МУКОПОЛИСАХАРИДОЗЫ
Под термином «мукополисахаридозы» объединяется ряд патологических
Наследственные болезни обмена веществ соединительной ткани.
МУКОПОЛИСАХАРИДОЗЫ
Под термином «мукополисахаридозы» объединяется ряд патологических
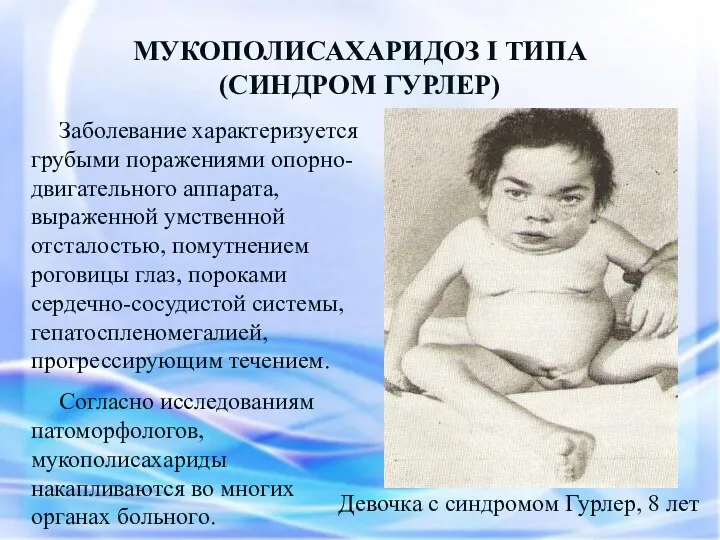
МУКОПОЛИСАХАРИДОЗ I ТИПА
(СИНДРОМ ГУРЛЕР)
Заболевание характеризуется грубыми поражениями опорно-двигательного аппарата, выраженной
МУКОПОЛИСАХАРИДОЗ I ТИПА
(СИНДРОМ ГУРЛЕР)
Заболевание характеризуется грубыми поражениями опорно-двигательного аппарата, выраженной
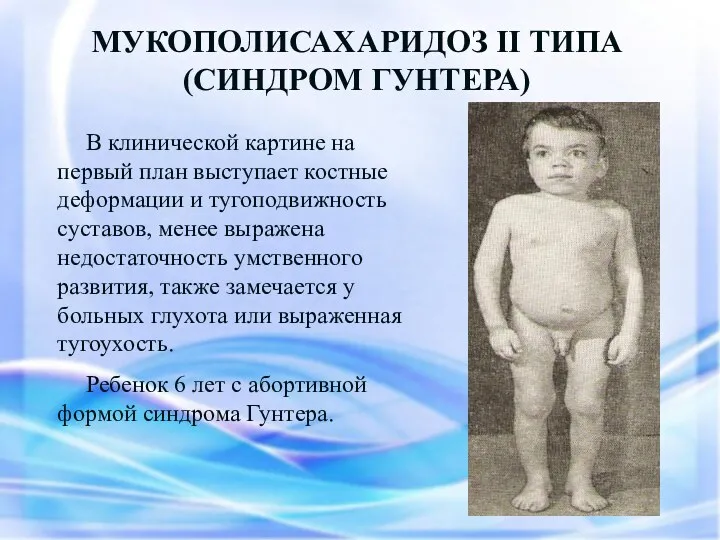
МУКОПОЛИСАХАРИДОЗ II ТИПА
(СИНДРОМ ГУНТЕРА)
В клинической картине на первый план выступает
МУКОПОЛИСАХАРИДОЗ II ТИПА
(СИНДРОМ ГУНТЕРА)
В клинической картине на первый план выступает
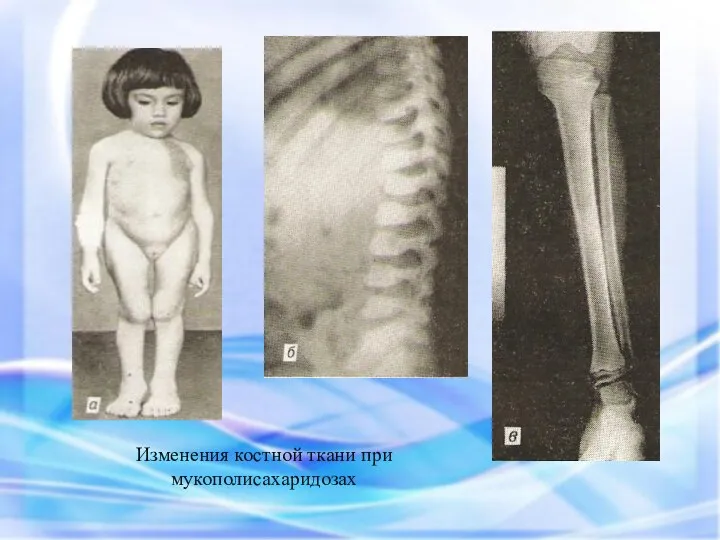
Изменения костной ткани при мукополисахаридозах
Изменения костной ткани при мукополисахаридозах
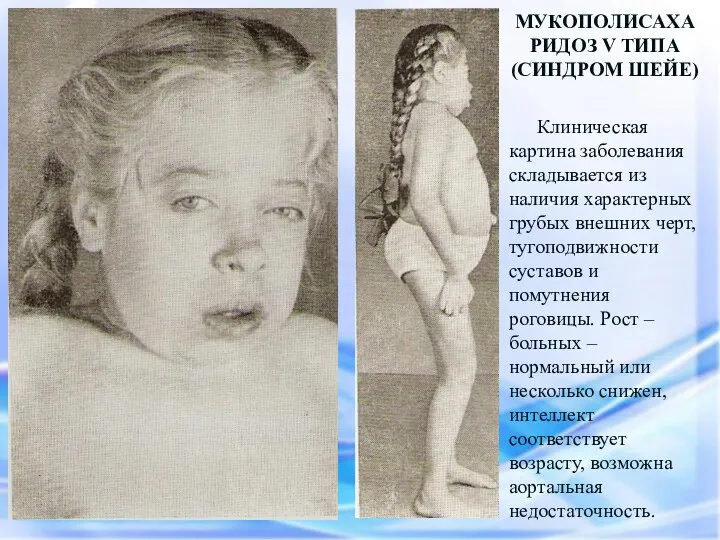
МУКОПОЛИСАХАРИДОЗ V ТИПА
(СИНДРОМ ШЕЙЕ)
Клиническая картина заболевания складывается из наличия
МУКОПОЛИСАХАРИДОЗ V ТИПА
(СИНДРОМ ШЕЙЕ)
Клиническая картина заболевания складывается из наличия
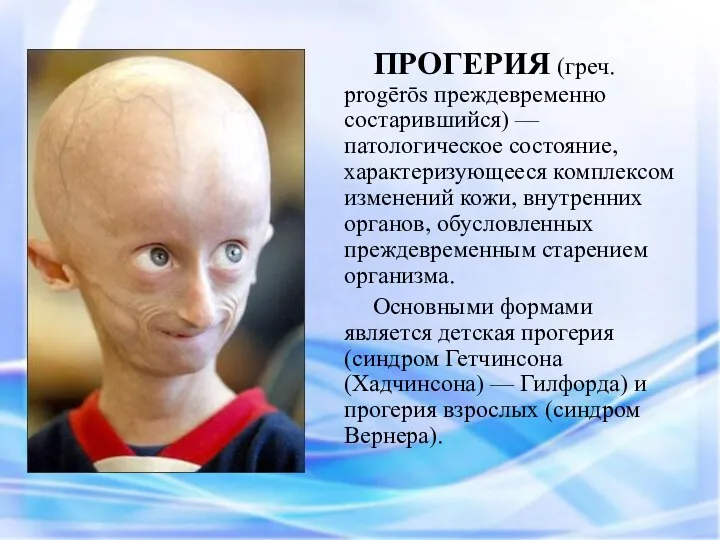
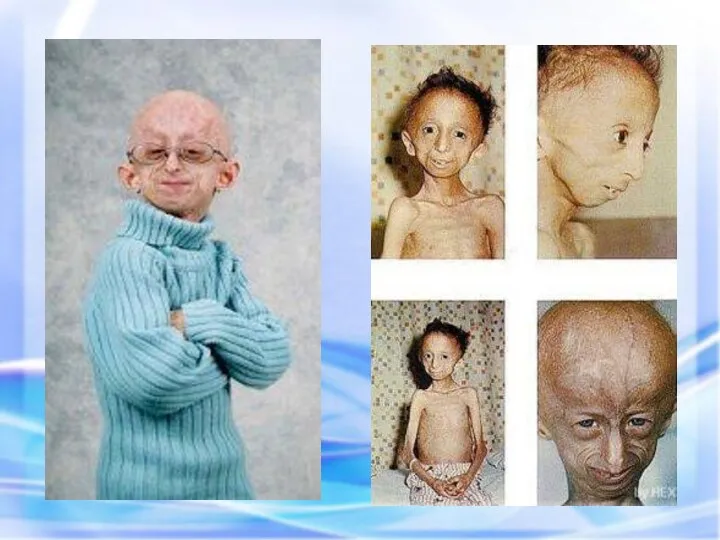
ПРОГЕРИЯ (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений
ПРОГЕРИЯ (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений
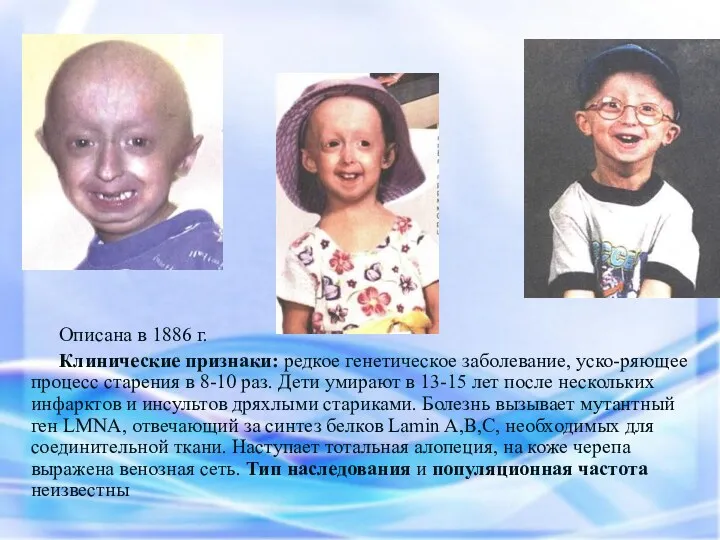
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения

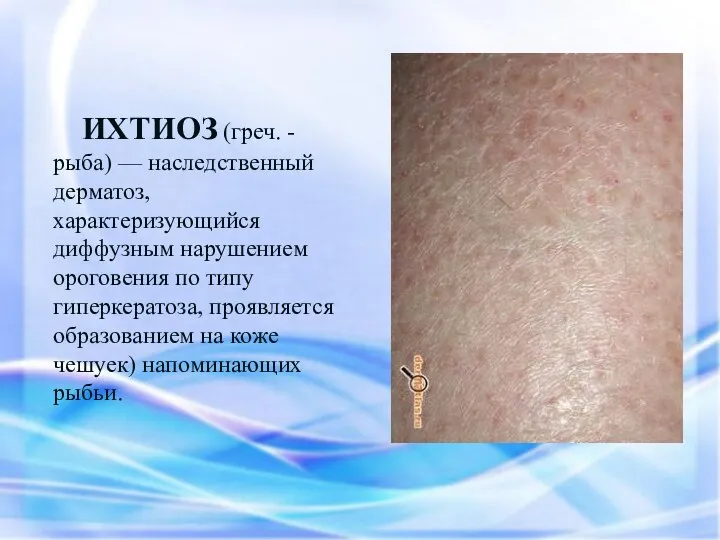
ИХТИОЗ (греч. - рыба) — наследственный дерматоз, характеризующийся диффузным нарушением ороговения по
ИХТИОЗ (греч. - рыба) — наследственный дерматоз, характеризующийся диффузным нарушением ороговения по
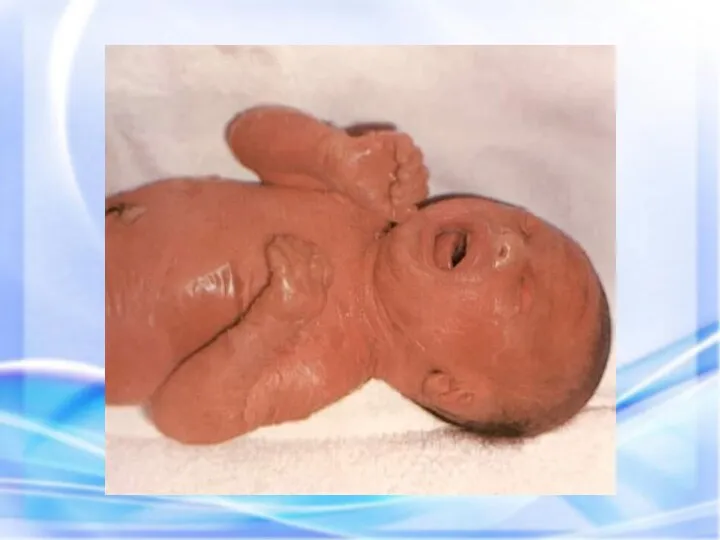
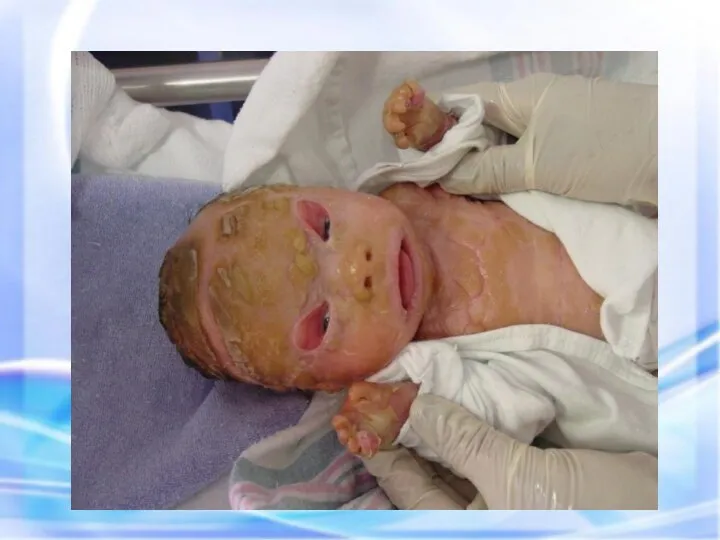
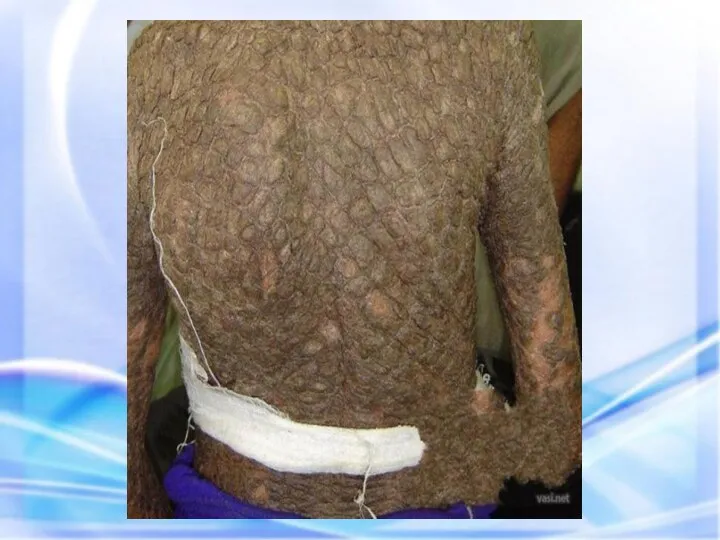

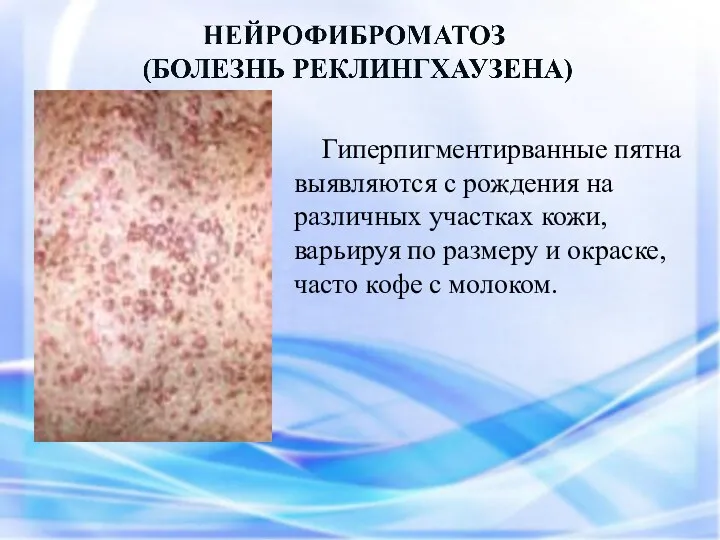
Гиперпигментирванные пятна выявляются с рождения на различных участках кожи, варьируя по
Гиперпигментирванные пятна выявляются с рождения на различных участках кожи, варьируя по
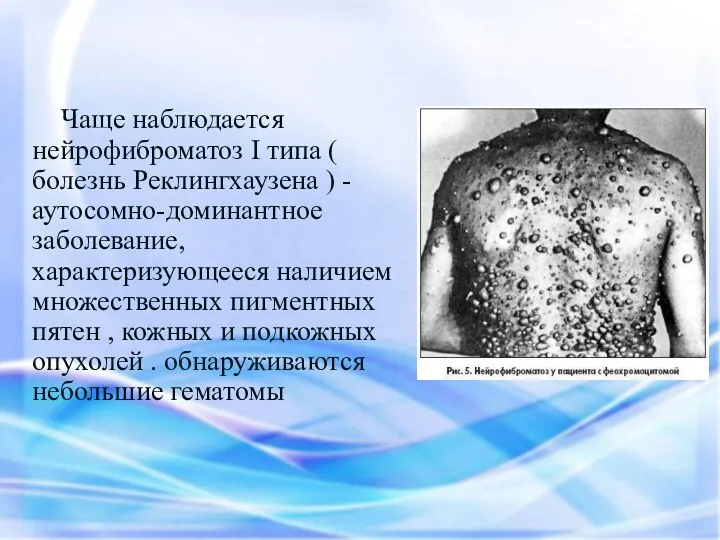
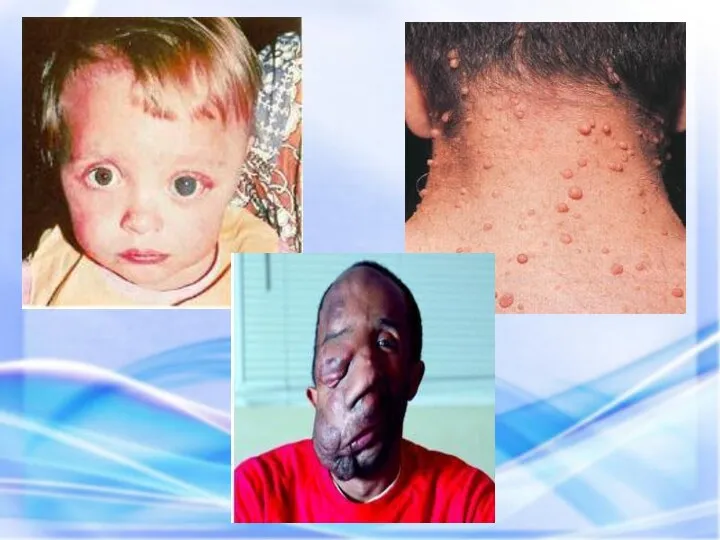
Чаще наблюдается нейрофиброматоз I типа ( болезнь Реклингхаузена ) - аутосомно-доминантное
Чаще наблюдается нейрофиброматоз I типа ( болезнь Реклингхаузена ) - аутосомно-доминантное
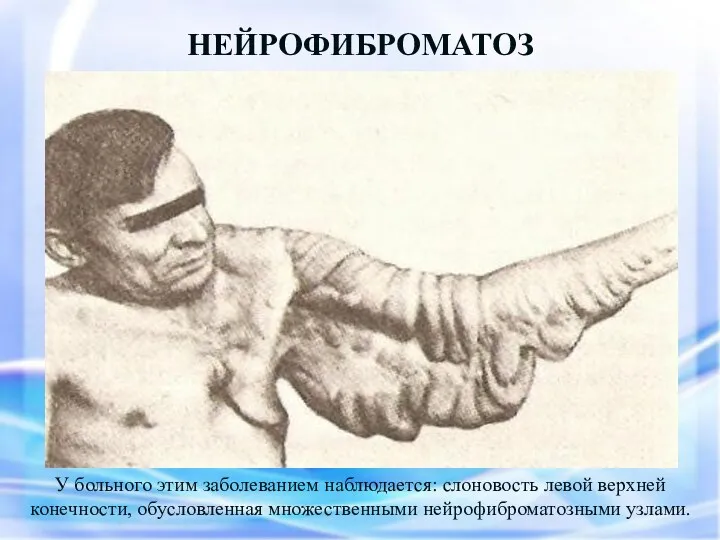
НЕЙРОФИБРОМАТОЗ
У больного этим заболеванием наблюдается: слоновость левой верхней конечности, обусловленная
НЕЙРОФИБРОМАТОЗ
У больного этим заболеванием наблюдается: слоновость левой верхней конечности, обусловленная
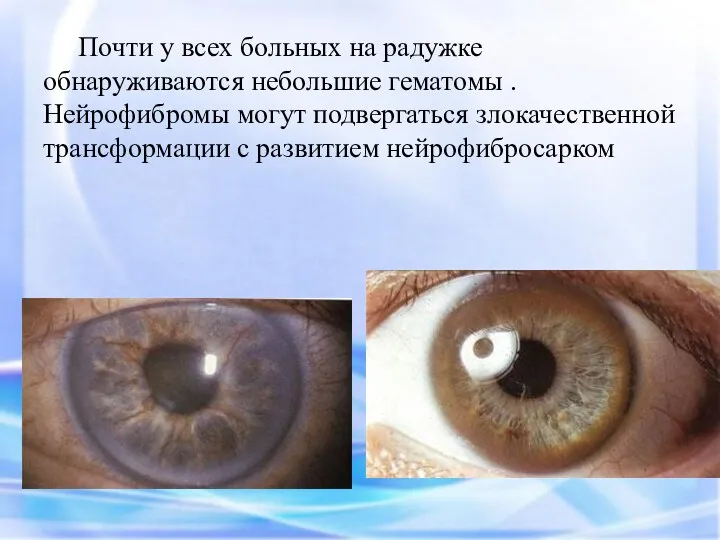
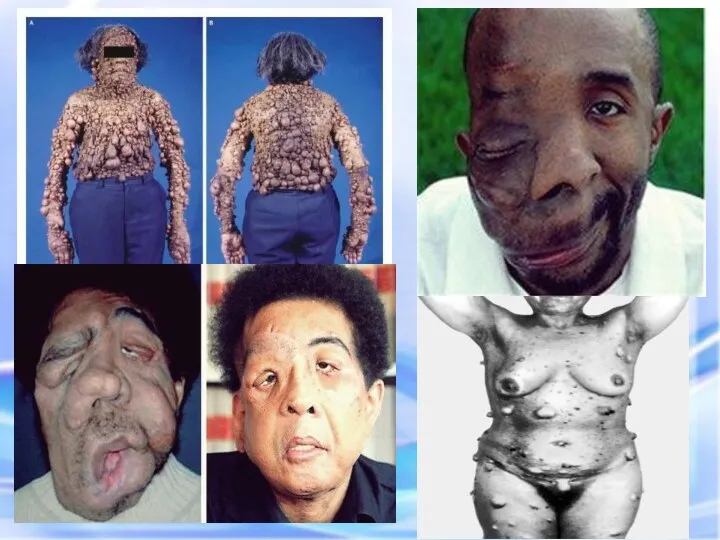
Почти у всех больных на радужке обнаруживаются небольшие гематомы . Нейрофибромы
Почти у всех больных на радужке обнаруживаются небольшие гематомы . Нейрофибромы
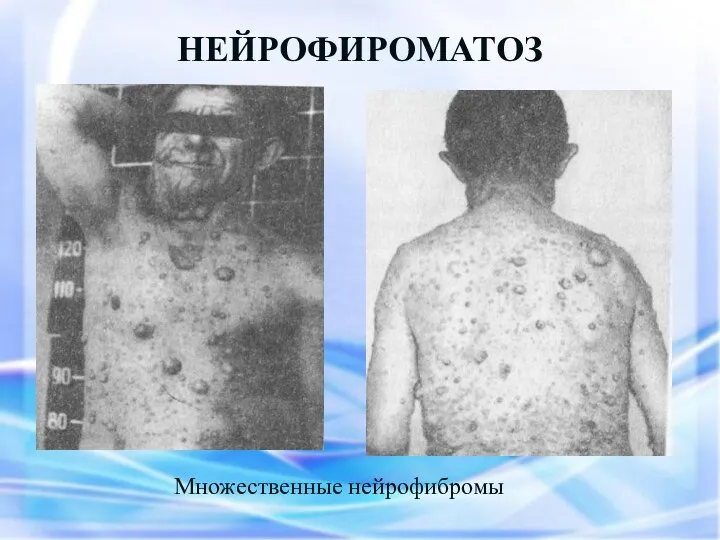
НЕЙРОФИРОМАТОЗ
Множественные нейрофибромы
НЕЙРОФИРОМАТОЗ
Множественные нейрофибромы


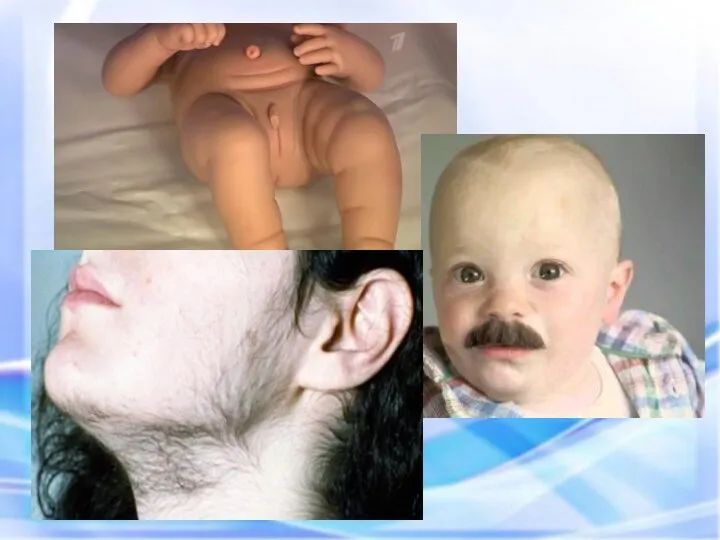
АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ (ВРАЖДЕННАЯ ГИПЕРПЛАЗИЯ КОРЫ НАДПОЧЕЧНИКОВ)
Адреногенитальный синдром (АГС) относится к группе
АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ (ВРАЖДЕННАЯ ГИПЕРПЛАЗИЯ КОРЫ НАДПОЧЕЧНИКОВ)
Адреногенитальный синдром (АГС) относится к группе

Новорожденная девочка с двойственным строением наружных половых органов
Мальчики шести лет (справа
Новорожденная девочка с двойственным строением наружных половых органов
Мальчики шести лет (справа


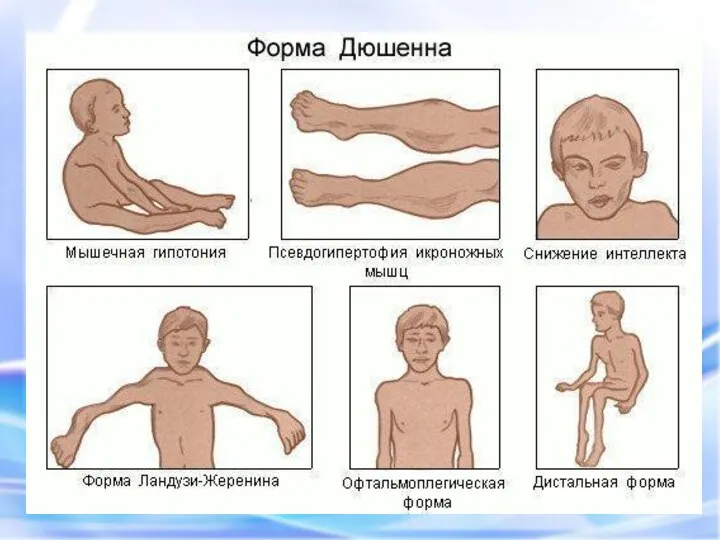
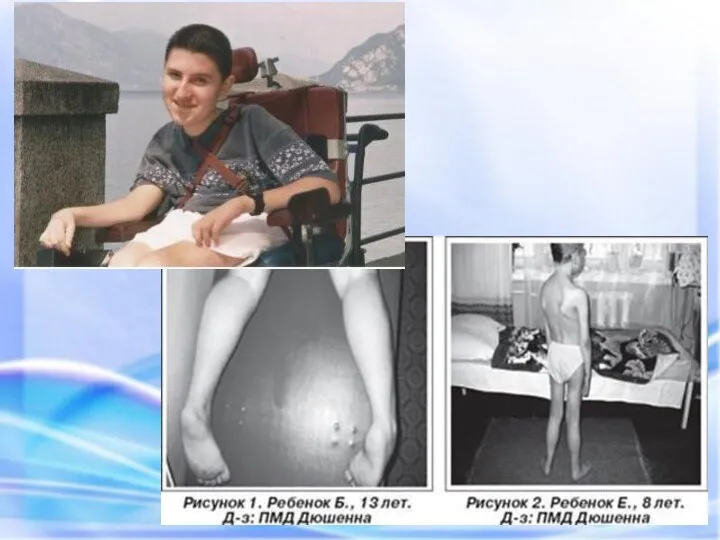
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МЫШЕЧНАЯ ДИСТРОФИЯ (ДЮШЕННА)
Это одна из самых частых форм наследственных нервно
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МЫШЕЧНАЯ ДИСТРОФИЯ (ДЮШЕННА)
Это одна из самых частых форм наследственных нервно
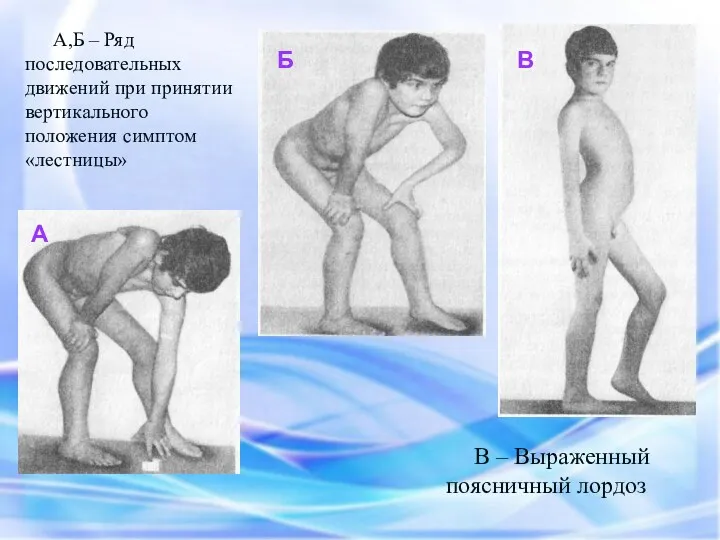
А,Б – Ряд последовательных движений при принятии вертикального положения симптом «лестницы»
А
Б
В
В
А,Б – Ряд последовательных движений при принятии вертикального положения симптом «лестницы»
А
Б
В
В

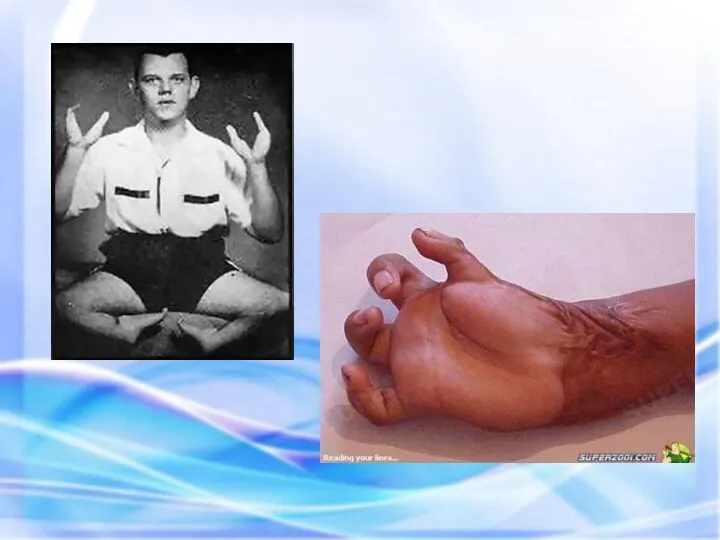
Странное племя людей-страусов (сапади) в Центральной Африке отличает от прочих обитателей
Странное племя людей-страусов (сапади) в Центральной Африке отличает от прочих обитателей
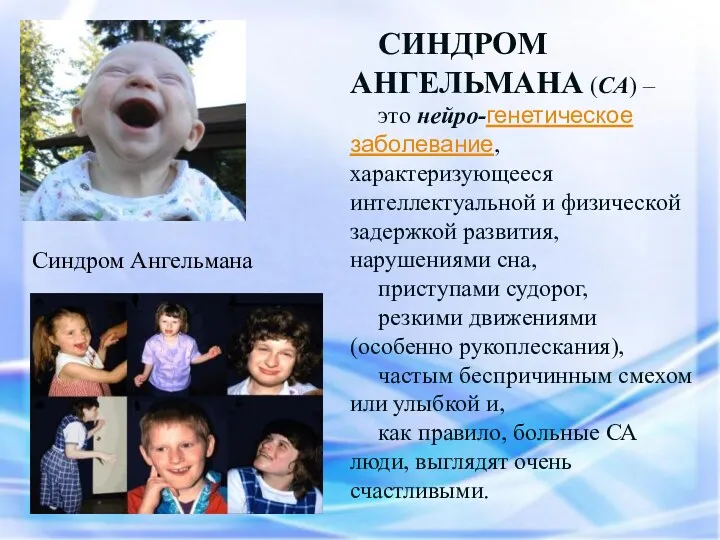
Синдром Ангельмана
СИНДРОМ АНГЕЛЬМАНА (СА) –
это нейро-генетическое заболевание, характеризующееся интеллектуальной и физической задержкой
Синдром Ангельмана
СИНДРОМ АНГЕЛЬМАНА (СА) –
это нейро-генетическое заболевание, характеризующееся интеллектуальной и физической задержкой
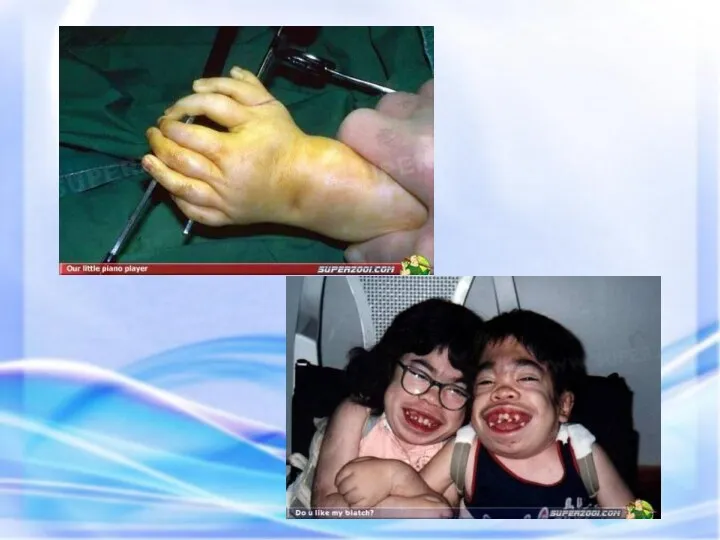


Дети наркоманов. Копия, воск.
Дети наркоманов. Копия, воск.
Сиамские близнецы, у родителей-наркоманов. Натура, заспиртованные.
Сиамские близнецы, у родителей-наркоманов. Натура, заспиртованные.

Дети у родителей больных наследственными заболеваниями. Копия, воск.
Дети у родителей больных наследственными заболеваниями. Копия, воск.
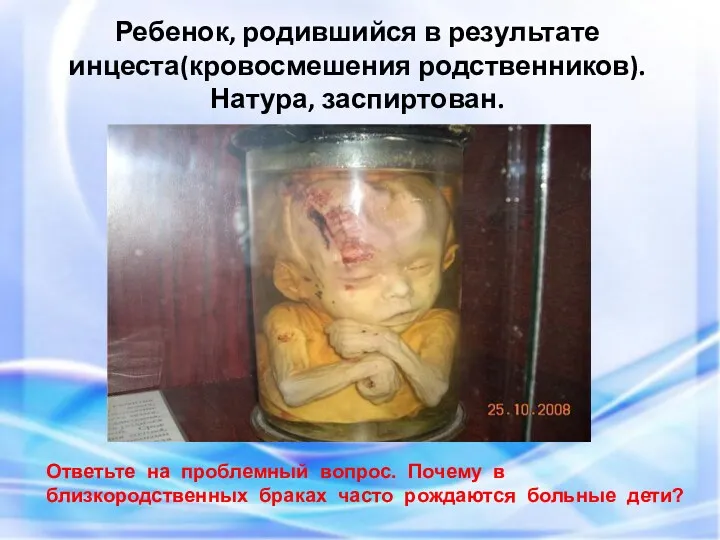
Ребенок, родившийся в результате
инцеста(кровосмешения родственников).
Натура, заспиртован.
Ответьте на проблемный вопрос.
Ребенок, родившийся в результате
инцеста(кровосмешения родственников).
Натура, заспиртован.
Ответьте на проблемный вопрос.

Ребенок, родившийся
в семье чернобыльцев. Натура, мумия.
Ребенок, родившийся
в семье чернобыльцев. Натура, мумия.

Человек-циклоп, и женщина-слон.
Жили в 19 веке. Копия, воск.
Человек-циклоп, и женщина-слон.
Жили в 19 веке. Копия, воск.
Согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом,
Согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом,

Если принять, что у человека примерно
100 000 генов и каждый
Если принять, что у человека примерно
100 000 генов и каждый

Вся история развития человека есть непрерывная цепь мутаций в нем. Мутации
Вся история развития человека есть непрерывная цепь мутаций в нем. Мутации
 Созылмалы лимфолейкоз
Созылмалы лимфолейкоз Первая помощь при инсульте и сердечной недостаточности
Первая помощь при инсульте и сердечной недостаточности Возбудитель холеры
Возбудитель холеры Формирование ЗОЖ у пожилых. Основы профилактики
Формирование ЗОЖ у пожилых. Основы профилактики Бронхтық демікпе
Бронхтық демікпе Лечение и профилактика туберкулеза
Лечение и профилактика туберкулеза prezentatsia_po_travme_2007
prezentatsia_po_travme_2007 Современные представления о заболеваниях пародонта. Классификации заболеваний пародонта. Клиника. Диагностика
Современные представления о заболеваниях пародонта. Классификации заболеваний пародонта. Клиника. Диагностика Профилактическая медицина
Профилактическая медицина Острый рассеянный энцефаломиелит
Острый рассеянный энцефаломиелит Организация производства экстракта полыни гмелина (Artemisia gmelini)
Организация производства экстракта полыни гмелина (Artemisia gmelini) Экзамен по гистологии
Экзамен по гистологии №16,17 Сложные реакции иммунитета
№16,17 Сложные реакции иммунитета Зәр шығару жүйесі
Зәр шығару жүйесі Сестринский уход при различных заболеваниях и состояниях. Лечение пациентов терапевтического профиля
Сестринский уход при различных заболеваниях и состояниях. Лечение пациентов терапевтического профиля Қайталап босанушы әйелдерде келесі жүктілікті қаламау мақсатында ЖІС мен гормональды контрацепцияның тиімділігі
Қайталап босанушы әйелдерде келесі жүктілікті қаламау мақсатында ЖІС мен гормональды контрацепцияның тиімділігі Хронические заболевания глотки
Хронические заболевания глотки Новости nCov-2019 (c 11/02 – COVID-2019, SARS CoV-2)
Новости nCov-2019 (c 11/02 – COVID-2019, SARS CoV-2) Геморагічні діатези
Геморагічні діатези Экстирпация матки
Экстирпация матки Вакцинадан кейінгі асқынулардың алдын алу,емдеу
Вакцинадан кейінгі асқынулардың алдын алу,емдеу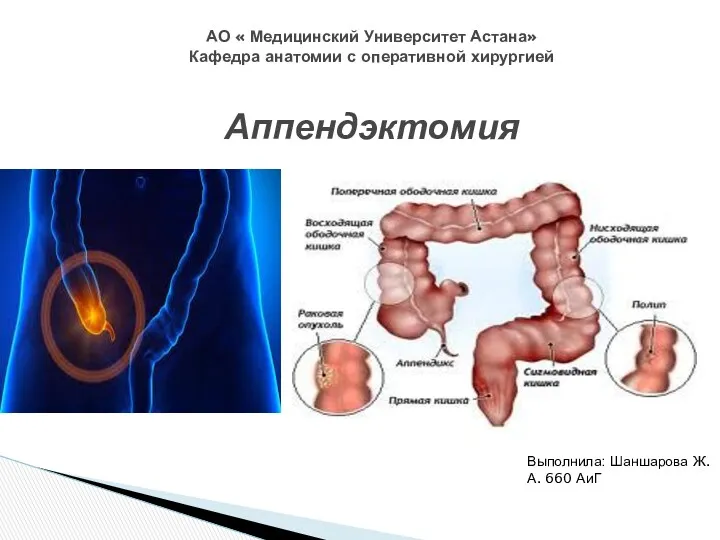 Топография слепой кишки
Топография слепой кишки Реабилитация больных, перенесших инсульт
Реабилитация больных, перенесших инсульт Питание детей старше года
Питание детей старше года Бронхиалды астманың ұстамасы кезіндегі алғашқы көмек
Бронхиалды астманың ұстамасы кезіндегі алғашқы көмек Анатомия и физиология мочевыделительной системы человека
Анатомия и физиология мочевыделительной системы человека Миокард инфаркті
Миокард инфаркті Эндокринді аурулардың лабораторлы диагностикасы
Эндокринді аурулардың лабораторлы диагностикасы