- Принципы дифференциальной диагностики коагулопатий

Содержание
- 2. План лекции «Принципы дифференциальной диагностики коагулопатий» 1.Понятие о геморрагических состояниях 2.Типы кровоточивости 3.Что такое гемофилия, разновидности
- 3. Приобретенные: Заболевания, в основе которых лежит патология одного или нескольких звеньев системы гемостаза и проявляющиеся геморрагическим
- 4. Методологические основы диагностики нарушений гемостаза Из клинических ориентиров наиболее важны следующие: Анамнез о давности заболевания, возможность
- 5. ТИПЫ КРОВОТОЧИВОСТИ
- 6. Васкулитно-пурпурный тип - объединяет геморрагии, обусловленные воспалительными изменениями микрососудов и периваскулярной ткани. Эти изменения чаще обусловлены
- 7. Основной вопрос при геморрагических проявлениях (микроциркуляторного типа): Являются ли кровотечения у больного следствием нарушений в системе
- 8. Клинико-патогенетическая классификация причин кровотечений I. Кровотечения, обусловленные местными деструктивно-некротическими процессами: новообразования, гранулемы и язвы, токсические и
- 9. Клинико-патогенетическая классификация причин кровотечений V. Кровотечения, обусловленные дефектами коагуляционного гемостаза и фибринолиза Врожденные коагулопатии (гемофилии А,
- 10. Гемофилии
- 11. Гемофилия А Геморрагическая коагулопатия, связанная со снижением активности фактора. Выделяют наследственную и приобретенную. Ген фактора VIII
- 12. Классификация гемофилии А
- 13. Гемофилия А Фактор VIII –антигемофильный глобулин А, циркулирует в крови в трех формах: коагулирующая единица (VIII
- 14. Ингибиторная форма гемофилии А Осложнение терапии препаратами фактора VIII. Проявляются у 5-20% с тяжелой формой гемофилии
- 15. Активность ингибитора Принцип метода основан на том, что при определении активности ингибитора, у него во время
- 16. Исследование активности ингибитора по методу Бетезда За 1 ЕД Бетезда принято такое количество антител, которое блокирует
- 17. Диагностический алгоритм После выявления удлинения АЧТВ и снижения активности фактора (VIII ) проводится тест смешивания. Плазма
- 18. Гемофилия В Встречается заболевание с частотой 1 на 30 000 случаев мужского населения. Причина меньшей частоты
- 19. Гемофилия В Кристмас- фактор, антигемофильный глобулин В, образуется в печени. Ген в Х хромосоме, в другом
- 20. Гемофилия С Активная форма образуется при участии фактора Х11. Врожденную недостаточность фактора Х1 называют болезнью Розенталя,
- 21. Дифференциация дефицита факторов VIII, IX, XI Результат при дефиците факторов
- 22. Выявление дефицита факторов с использованием принципа заменных проб Например, АЧТВ удлинено Плазма без фактора VIII 0,5
- 23. Дефицит факторов II, V, VII, X. При одновременном удлинении времени свертывания в протромбиновом времени и АЧТВ-тестах
- 24. Дифференциация дефицита факторов VII, V, X, II протромбинового комплекса в тестах с гетерогенными коагулазами
- 25. Дефицит факторов VII и X Врожденный дефицит фактора VII (гипопроконвертинемия) передается как аутосомно-рецессивный признак. Клинических проявлений
- 26. Кровотечения, возникающие у больных после операций на предстательной железе, при язвенном поражении ЖКТ, при кровоизлияниях в
- 27. Заболевание наследуется по аутосомно-доминантному типу. Проявляется у обоих полов, с большей частотой у женщин (59%) По
- 28. Болезнь Виллебранда Причина кровотечений 1. Нарушение свертываемости крови из-за снижения активности или отсутствия фактора Виллебранда (обеспечивает
- 29. ПРИОБРЕТЕННЫЙ СИНДРОМ Болезнь Виллебранда определяется у пациентов с аутоиммунными, лимфопролиферативными заболеваниями, обусловлен появлением ингибитора против фактора
- 30. Все о факторе Виллебранда Синтезируется в эндотелиоцитах и мегакариоцитах, Т1/2 – 9-15 часов; Хранится в тельцах
- 31. Варианты взаимодействия фактораVIII
- 32. 3 типа болезни Виллебранда: https://www.pinterest.com/.../ 469570698623449.
- 33. Классификация болезни Виллебранда 1-й тип обусловлен частичным количественным дефицитом фактора Виллебранда снижение прокоагулянтной активности фактора VIII
- 34. Классификация болезни Виллебранда 3-й тип тяжелая форма с полным дефицитом фактора Виллебранда. форма характеризуется отсутствием фактора
- 35. Основные подтипы болезни Виллебранда
- 36. Клиническая картина Кровотечения из слизистых полости рта, носа, внутренних органов. Симптомы кровоточивости варьируют от умеренно выраженных
- 37. Лабораторная диагностика болезни Виллебранда Скрининговые тесты Время кровотечения - удлинено не более, чем у 50% больных,
- 38. График зависимости оптической плотности (осьY) от времени (ось Х). Зеленым цветом выделена кривая, соответствующая нормальной плазме,
- 39. Ристоцетин-кофакторная активность vWF:RCo Принцип метода: Специально приготовленные контрольные тромбоциты инкубируются с ристоцетином. Без участия ф.Виллебранда тромбоциты
- 40. Тест с ристоцетином для выяснения подтипа болезни Виллебранда Если результат теста «Ристоцетин кофактор» у больного дал
- 41. Определение мультимерности ФВ (иммуноэлектрофорез) Коллаген связывающая активность vWF:CB Иммуноферментный метод определения адгезивных свойств ФВ «Скрининговый» функциональный
- 42. Лабораторная диагностика Отношения: vWF:RCo/ vWF:Ag FVIII/vWF:Ag
- 43. Возможный диагностический алгоритм VWF:Ag Имеется Отсутствует Тип 3 Определение VWF:RCo VWF:RCo VWF:Ag >0,7 Тип 1 Тип
- 44. Норма Норма Норма Норма Норма Дифференциальный диагноз форм болезни Виллебранда
- 45. Женщина 30 лет, мама ребенка больного болезнью Виллебранда. Пришла на прием к гематологу с целью исключения
- 46. Лабораторная оценка гемостаза при кровоточивости MAYO Laboratories, 2009, USA
- 48. Скачать презентацию

План лекции «Принципы дифференциальной диагностики коагулопатий»
1.Понятие о геморрагических состояниях
2.Типы кровоточивости
3.Что такое
План лекции «Принципы дифференциальной диагностики коагулопатий»
1.Понятие о геморрагических состояниях
2.Типы кровоточивости
3.Что такое
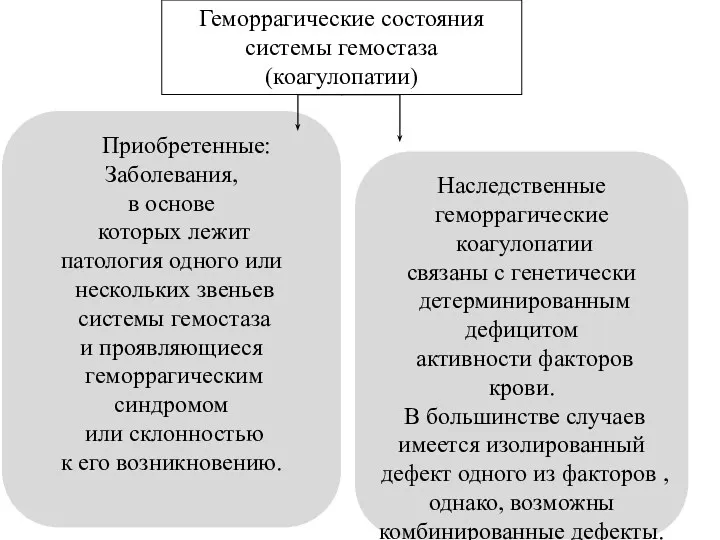
Приобретенные:
Заболевания,
в основе
которых лежит
патология одного или
нескольких
Приобретенные:
Заболевания,
в основе
которых лежит
патология одного или
нескольких
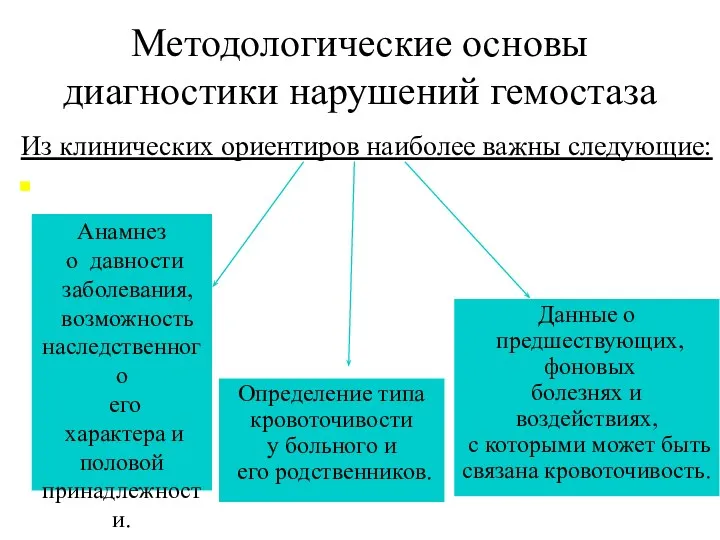
Методологические основы диагностики нарушений гемостаза
Из клинических ориентиров наиболее важны следующие:
Анамнез
Методологические основы диагностики нарушений гемостаза
Из клинических ориентиров наиболее важны следующие:
Анамнез

ТИПЫ КРОВОТОЧИВОСТИ
ТИПЫ КРОВОТОЧИВОСТИ

Васкулитно-пурпурный тип - объединяет геморрагии, обусловленные воспалительными изменениями микрососудов и периваскулярной
Васкулитно-пурпурный тип - объединяет геморрагии, обусловленные воспалительными изменениями микрососудов и периваскулярной

Основной вопрос при геморрагических проявлениях (микроциркуляторного типа): Являются ли кровотечения у
Основной вопрос при геморрагических проявлениях (микроциркуляторного типа): Являются ли кровотечения у

Клинико-патогенетическая классификация
причин кровотечений
I. Кровотечения, обусловленные местными деструктивно-некротическими процессами: новообразования, гранулемы
Клинико-патогенетическая классификация
причин кровотечений
I. Кровотечения, обусловленные местными деструктивно-некротическими процессами: новообразования, гранулемы

Клинико-патогенетическая классификация причин кровотечений
V. Кровотечения, обусловленные дефектами коагуляционного гемостаза и фибринолиза
Врожденные
Клинико-патогенетическая классификация причин кровотечений
V. Кровотечения, обусловленные дефектами коагуляционного гемостаза и фибринолиза
Врожденные
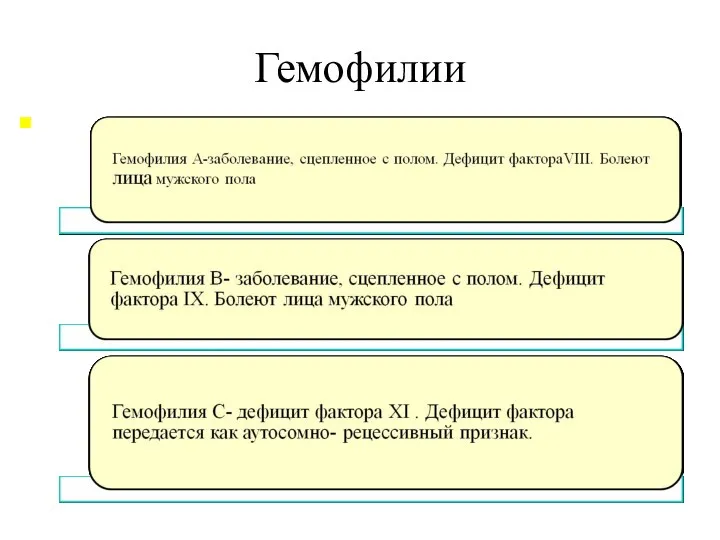
Гемофилии
Гемофилии
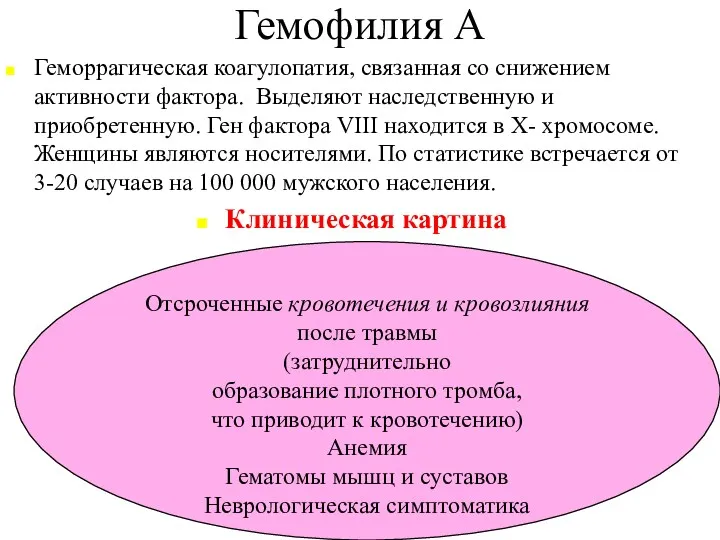
Гемофилия А
Геморрагическая коагулопатия, связанная со снижением активности фактора. Выделяют наследственную и
Гемофилия А
Геморрагическая коагулопатия, связанная со снижением активности фактора. Выделяют наследственную и
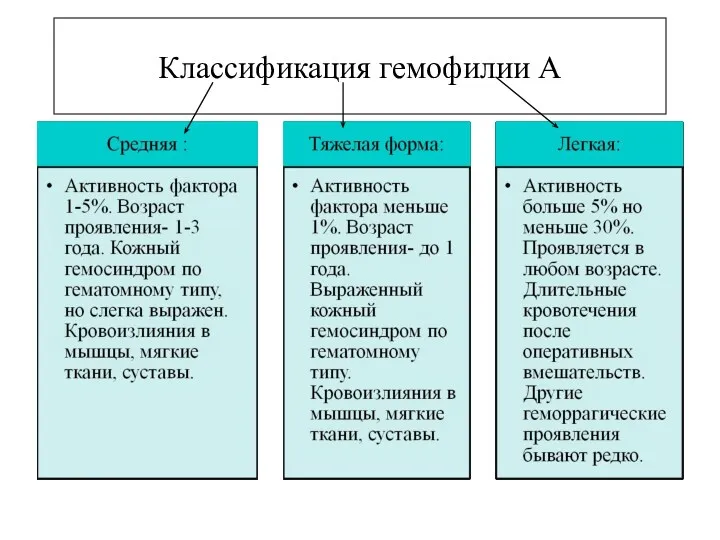
Классификация гемофилии А
Классификация гемофилии А

Гемофилия А
Фактор VIII –антигемофильный глобулин А, циркулирует в крови в трех
Гемофилия А
Фактор VIII –антигемофильный глобулин А, циркулирует в крови в трех

Ингибиторная форма гемофилии А
Осложнение терапии препаратами
фактора VIII.
Проявляются у 5-20%
Ингибиторная форма гемофилии А
Осложнение терапии препаратами
фактора VIII.
Проявляются у 5-20%

Активность ингибитора
Принцип метода основан на том, что при определении активности ингибитора,
Активность ингибитора
Принцип метода основан на том, что при определении активности ингибитора,

Исследование активности ингибитора по методу Бетезда
За 1 ЕД Бетезда принято такое
Исследование активности ингибитора по методу Бетезда
За 1 ЕД Бетезда принято такое

Диагностический алгоритм
После выявления удлинения АЧТВ и снижения активности фактора (VIII )
Диагностический алгоритм
После выявления удлинения АЧТВ и снижения активности фактора (VIII )
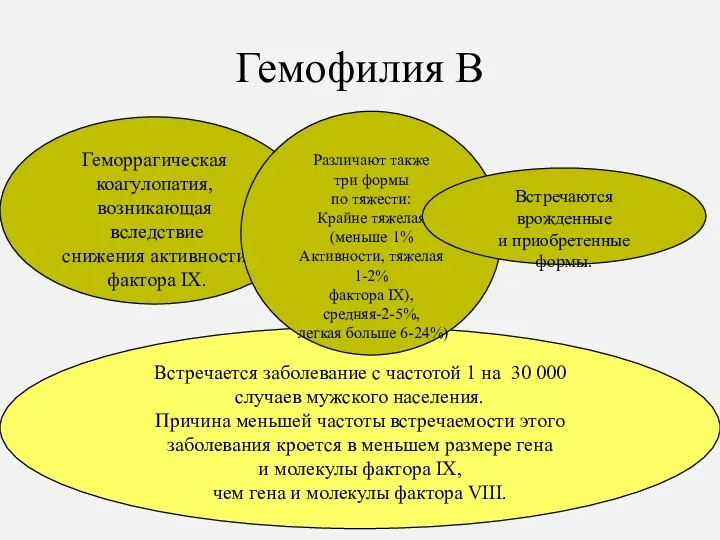
Гемофилия В
Встречается заболевание с частотой 1 на 30 000
случаев мужского
Гемофилия В
Встречается заболевание с частотой 1 на 30 000
случаев мужского

Гемофилия В
Кристмас- фактор, антигемофильный глобулин В, образуется в печени. Ген в
Гемофилия В
Кристмас- фактор, антигемофильный глобулин В, образуется в печени. Ген в

Гемофилия С
Активная форма образуется при участии фактора Х11. Врожденную недостаточность фактора
Гемофилия С
Активная форма образуется при участии фактора Х11. Врожденную недостаточность фактора
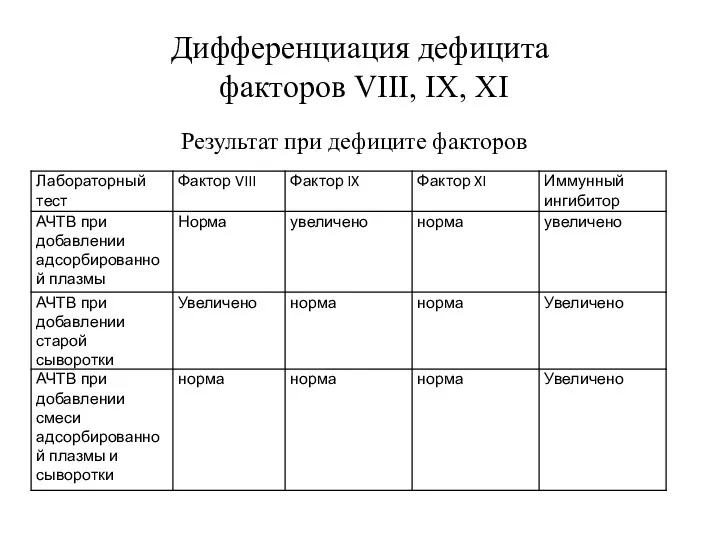
Дифференциация дефицита
факторов VIII, IX, XI
Результат при дефиците факторов
Дифференциация дефицита
факторов VIII, IX, XI
Результат при дефиците факторов
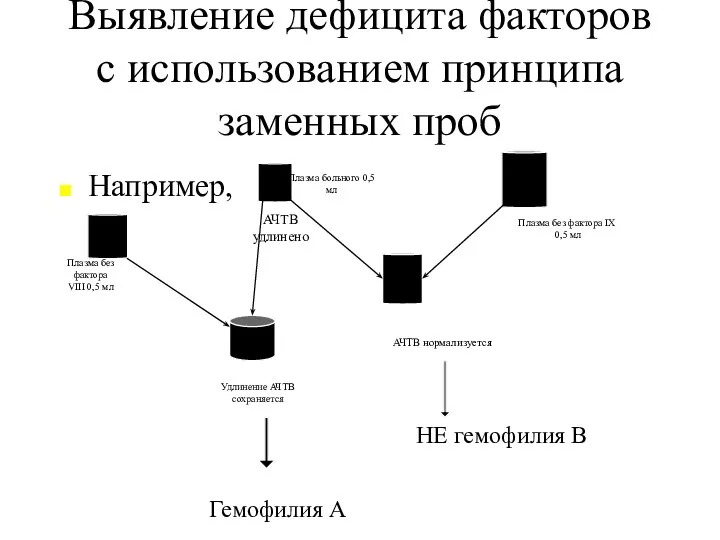
Выявление дефицита факторов с использованием принципа заменных проб
Например,
АЧТВ удлинено
Плазма без
Выявление дефицита факторов с использованием принципа заменных проб
Например,
АЧТВ удлинено
Плазма без

Дефицит факторов II, V, VII, X.
При одновременном удлинении времени свертывания в
Дефицит факторов II, V, VII, X.
При одновременном удлинении времени свертывания в

Дифференциация дефицита факторов VII, V, X, II протромбинового комплекса в тестах
Дифференциация дефицита факторов VII, V, X, II протромбинового комплекса в тестах

Дефицит факторов VII и X
Врожденный дефицит фактора VII (гипопроконвертинемия) передается как
Дефицит факторов VII и X
Врожденный дефицит фактора VII (гипопроконвертинемия) передается как

Кровотечения, возникающие у больных после операций на предстательной железе, при язвенном
Кровотечения, возникающие у больных после операций на предстательной железе, при язвенном
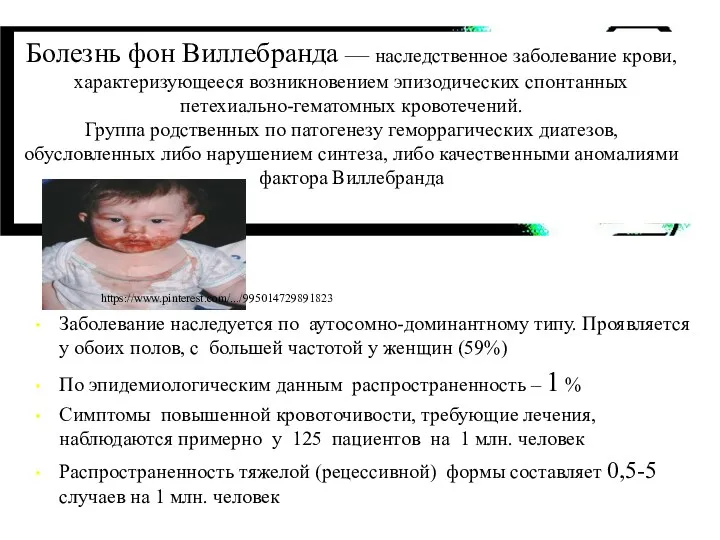
Заболевание наследуется по аутосомно-доминантному типу. Проявляется у обоих полов, с большей
Заболевание наследуется по аутосомно-доминантному типу. Проявляется у обоих полов, с большей

Болезнь Виллебранда
Причина кровотечений
1. Нарушение свертываемости крови из-за снижения активности или отсутствия фактора
Болезнь Виллебранда
Причина кровотечений
1. Нарушение свертываемости крови из-за снижения активности или отсутствия фактора

ПРИОБРЕТЕННЫЙ СИНДРОМ
Болезнь Виллебранда определяется у пациентов с аутоиммунными, лимфопролиферативными заболеваниями,
обусловлен появлением
ПРИОБРЕТЕННЫЙ СИНДРОМ
Болезнь Виллебранда определяется у пациентов с аутоиммунными, лимфопролиферативными заболеваниями, обусловлен появлением

Все о факторе Виллебранда
Синтезируется в эндотелиоцитах и мегакариоцитах,
Т1/2 –
Все о факторе Виллебранда
Синтезируется в эндотелиоцитах и мегакариоцитах,
Т1/2 –

Варианты взаимодействия фактораVIII
Варианты взаимодействия фактораVIII
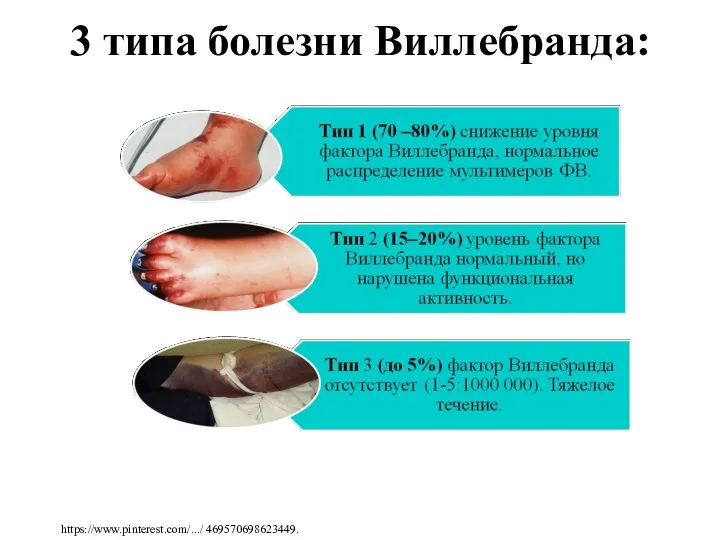
3 типа болезни Виллебранда:
https://www.pinterest.com/.../ 469570698623449.
3 типа болезни Виллебранда:
https://www.pinterest.com/.../ 469570698623449.

Классификация болезни Виллебранда
1-й тип
обусловлен частичным количественным дефицитом фактора Виллебранда
снижение прокоагулянтной активности
Классификация болезни Виллебранда
1-й тип
обусловлен частичным количественным дефицитом фактора Виллебранда
снижение прокоагулянтной активности

Классификация болезни Виллебранда
3-й тип
тяжелая форма с полным дефицитом фактора Виллебранда.
Классификация болезни Виллебранда
3-й тип
тяжелая форма с полным дефицитом фактора Виллебранда.

Основные подтипы болезни Виллебранда
Основные подтипы болезни Виллебранда

Клиническая картина
Кровотечения из слизистых полости рта, носа, внутренних органов.
Симптомы кровоточивости
Клиническая картина
Кровотечения из слизистых полости рта, носа, внутренних органов.
Симптомы кровоточивости

Лабораторная диагностика болезни Виллебранда
Скрининговые тесты
Время кровотечения - удлинено не более, чем
Лабораторная диагностика болезни Виллебранда
Скрининговые тесты
Время кровотечения - удлинено не более, чем
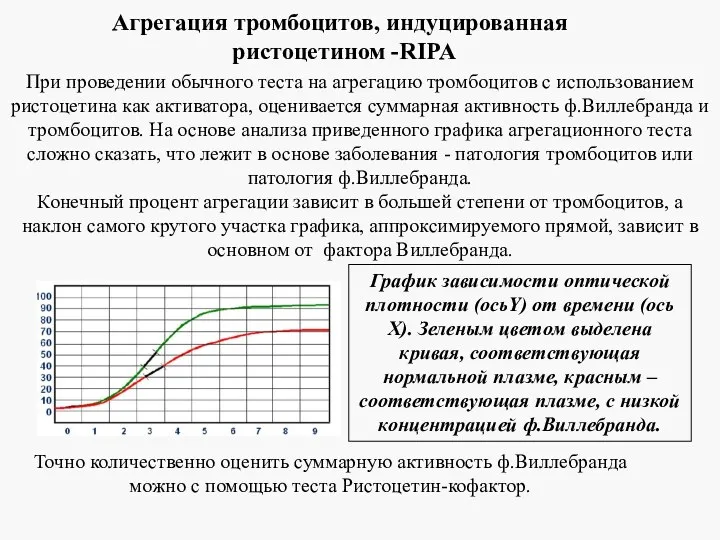
График зависимости оптической плотности (осьY) от времени (ось Х). Зеленым цветом
График зависимости оптической плотности (осьY) от времени (ось Х). Зеленым цветом
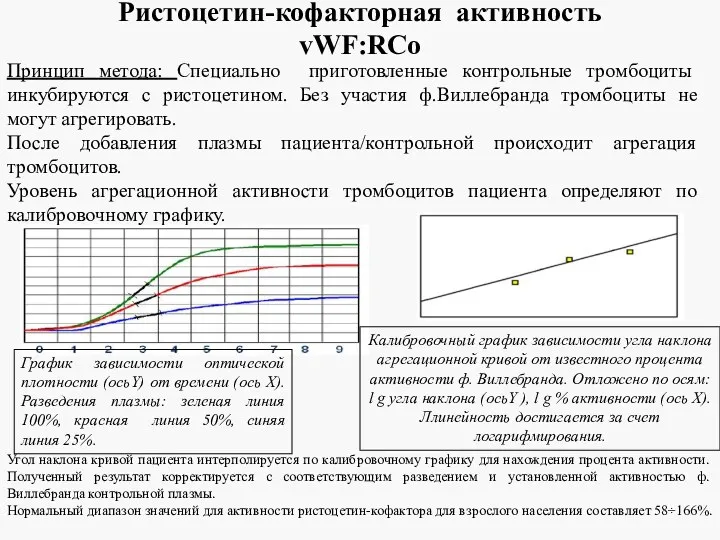
Ристоцетин-кофакторная активность vWF:RCo
Принцип метода: Специально приготовленные контрольные тромбоциты инкубируются с
Ристоцетин-кофакторная активность vWF:RCo
Принцип метода: Специально приготовленные контрольные тромбоциты инкубируются с
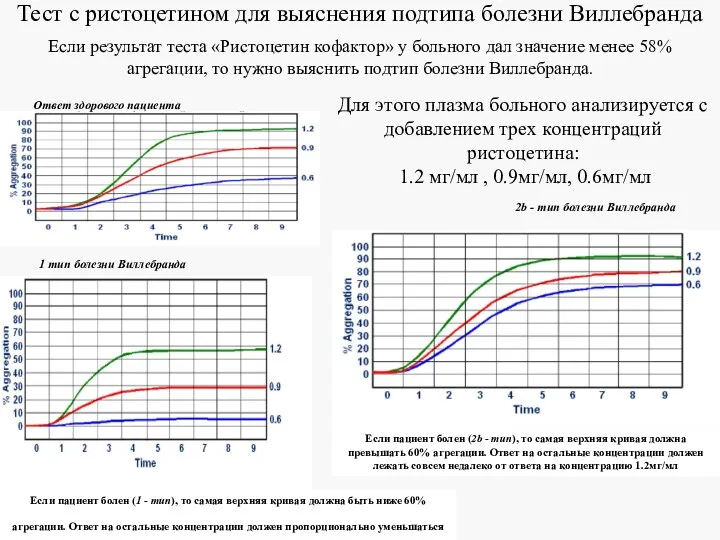
Тест с ристоцетином для выяснения подтипа болезни Виллебранда
Если результат теста «Ристоцетин
Тест с ристоцетином для выяснения подтипа болезни Виллебранда
Если результат теста «Ристоцетин

Определение мультимерности ФВ (иммуноэлектрофорез)
Коллаген связывающая активность vWF:CB
Иммуноферментный метод определения адгезивных свойств
Определение мультимерности ФВ (иммуноэлектрофорез)
Коллаген связывающая активность vWF:CB
Иммуноферментный метод определения адгезивных свойств

Лабораторная диагностика
Отношения: vWF:RCo/ vWF:Ag FVIII/vWF:Ag
Лабораторная диагностика
Отношения: vWF:RCo/ vWF:Ag FVIII/vWF:Ag
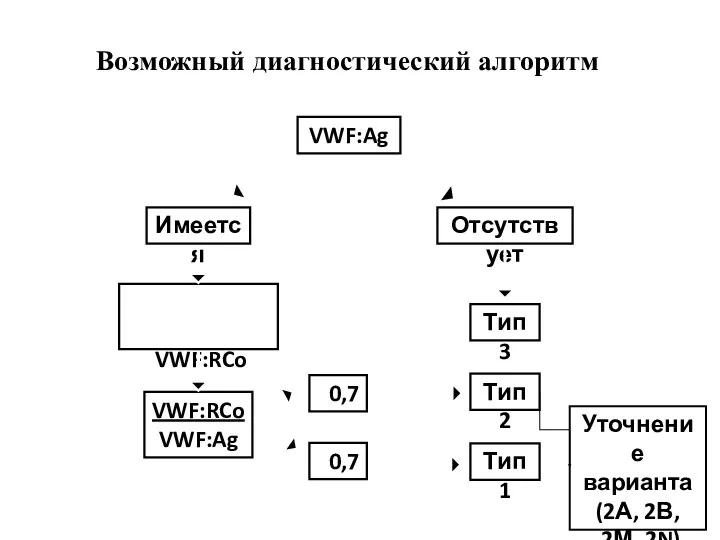
Возможный диагностический алгоритм
VWF:Ag
Имеется
Отсутствует
Тип 3
Определение
VWF:RCo
VWF:RCo
VWF:Ag
>0,7
<0,7
Тип 1
Тип 2
Уточнение
варианта
(2А, 2В,
2М, 2N)
Возможный диагностический алгоритм
VWF:Ag
Имеется
Отсутствует
Тип 3
Определение
VWF:RCo
VWF:RCo
VWF:Ag
>0,7
<0,7
Тип 1
Тип 2
Уточнение
варианта
(2А, 2В,
2М, 2N)
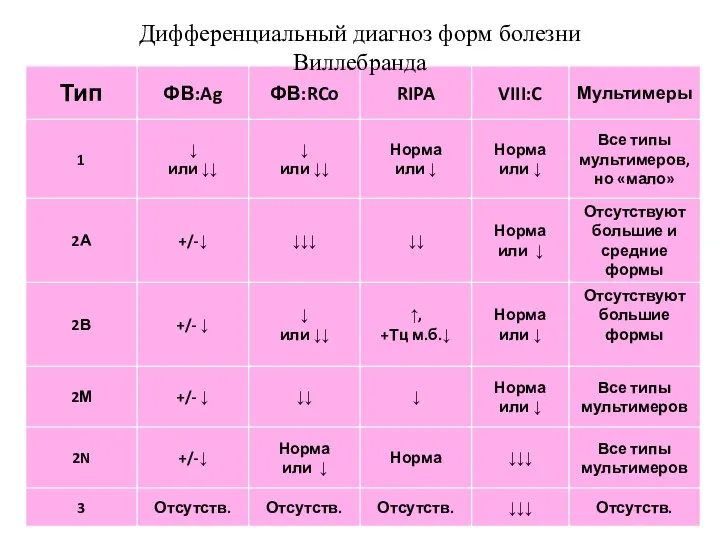
Норма
Норма
Норма
Норма
Норма
Дифференциальный диагноз форм болезни Виллебранда
Норма
Норма
Норма
Норма
Норма
Дифференциальный диагноз форм болезни Виллебранда

Женщина 30 лет, мама ребенка больного болезнью Виллебранда. Пришла на прием
Женщина 30 лет, мама ребенка больного болезнью Виллебранда. Пришла на прием
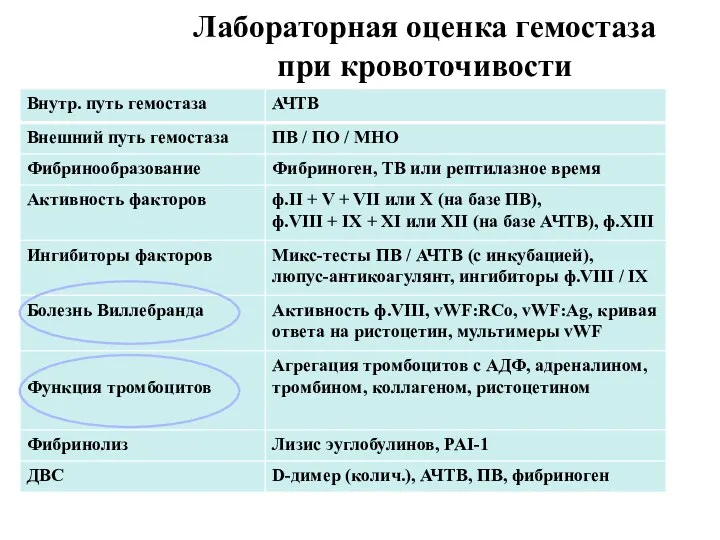
Лабораторная оценка гемостаза
при кровоточивости
MAYO Laboratories, 2009, USA
Лабораторная оценка гемостаза
при кровоточивости
MAYO Laboratories, 2009, USA
 Клиникалық эпидемиологиядағы биологиялық статистиканың рөлі
Клиникалық эпидемиологиядағы биологиялық статистиканың рөлі ГБУЗ Городская больница №2 г.Волжский
ГБУЗ Городская больница №2 г.Волжский Воспалительные заболевания челюстно-лицевой области
Воспалительные заболевания челюстно-лицевой области Дизентерия. Сальмонеллез. Тырысқақ
Дизентерия. Сальмонеллез. Тырысқақ Фотодинамическая терапия
Фотодинамическая терапия Естественное вскармливание. Преимущества. Питание, режим и гигиена кормящей матери
Естественное вскармливание. Преимущества. Питание, режим и гигиена кормящей матери Антибактериальная терапия респираторных инфекций с позиций доказательной медицины
Антибактериальная терапия респираторных инфекций с позиций доказательной медицины Деформации шеи
Деформации шеи Гипоксия
Гипоксия Босанудың түрлі аномалияларындағы партограмма толтыру ережесі
Босанудың түрлі аномалияларындағы партограмма толтыру ережесі Неотложная помощь при бронхообструктивном синдроме
Неотложная помощь при бронхообструктивном синдроме Сахарный диабет
Сахарный диабет Рубцы. Этиология
Рубцы. Этиология Питание в пожилом и старческом возрасте
Питание в пожилом и старческом возрасте Артериальная гипертензия
Артериальная гипертензия Захворювання серцево-судинної системи у дітей. Лекція 9
Захворювання серцево-судинної системи у дітей. Лекція 9 Паталогическая анатомия в комиксах и мемах. Тема: Некроз. Апоптоз. Дистрофии (паренхиматозные)
Паталогическая анатомия в комиксах и мемах. Тема: Некроз. Апоптоз. Дистрофии (паренхиматозные) Пневмонии. Респираторный отдел
Пневмонии. Респираторный отдел Лекарственные средства, влияющие на афферентную нервную систему
Лекарственные средства, влияющие на афферентную нервную систему Роль лучевых методов исследования в диагностике профессиональных заболеваний бронхолёгочной системы
Роль лучевых методов исследования в диагностике профессиональных заболеваний бронхолёгочной системы Балалардағы аритмиялар
Балалардағы аритмиялар Проведение профилактических медицинских осмотров целевых групп населения
Проведение профилактических медицинских осмотров целевых групп населения Онкоурология. Рак почки
Онкоурология. Рак почки Лечение инфаркта миокарда
Лечение инфаркта миокарда ЛЕКЦИЯ ВГ_
ЛЕКЦИЯ ВГ_ День медицинского работника
День медицинского работника Виды повязок
Виды повязок Неотложные состояния в пульмонологии и гематологии
Неотложные состояния в пульмонологии и гематологии