- Прогрессирующие мышечные дистрофии Дюшенна и Бекера

Содержание
- 2. Мышечная дистрофия Дюшенна. -прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением
- 3. Генетическое нарушение. Патология появляется вследствие генной мутации, имеющей место в области хр21. Более четверти таких патологий
- 4. Тип наследования. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что
- 5. Частота заболевания. Распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков.
- 6. Возраст манифестации. Миодистрофия Дюшенна характеризуется началом в первые 1-5 лет жизни ребенка, тяжелым течением, приводящим к
- 7. Основные клинические проявления Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка.
- 8. Основные клинические проявления Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной
- 9. Симптом Говерса.
- 10. Другие типичные симптомы
- 11. Основные клинические проявления Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно
- 12. Основные клинические проявления Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз,
- 13. Основные клинические проявления. Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12
- 14. Диагностика Установить диагноз миодистрофии Дюшенна помогают: анамнез неврологическое обследование результаты электрофизиологического тестирования определение креатинфосфокиназы (КФК) в
- 15. Диагностика Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует
- 16. Диагностика В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана
- 17. Специфическое лечение Специфического лечения в настоящее время не существует. Лечение только симтоматическое: Для улучшения метаболизма мышечной
- 18. Справка. В норме белок дистрофин поддерживает целостность сарколеммы - мембраны миоцитов (мышечных волокон), обеспечивает эластичность и
- 19. Мышечная дистрофия Беккера. вариант наследственной сцепленной с Х-хромосомой миодистрофии, отличающейся более замедленным и доброкачественным течением. Заболевание
- 20. Генетическое нарушение В основе заболевания лежит мутация в гене, ответственном за кодирование белка дистрофина. Примерно 30%
- 21. Тип наследования и частота заболевания. Патология наследуется рецессивно сцеплено с Х-хромосомой, поэтому болеют только лица мужского
- 22. Возраст манифестации. Прогрессирующая мышечная дистрофия Беккера манифестирует обычно в период от 10 до 15 лет, в
- 23. Основные клинические симптомы. Начальными признаками заболевания выступают чрезмерная утомляемость и мышечная слабость в тазовом поясе и
- 24. Основные клинические симптомы. Как и другие наследственные миопатии, заболевание Беккера характеризуется симметрично развивающимися атрофиями мышц. В
- 25. Основные клинические симптомы Клиническая картина мышечной дистрофии Беккера во многом сходна с миодистрофией Дюшенна. Усугубление мышечной
- 28. Диагностика. Прогрессирующая мышечная дистрофия Беккера диагностируется неврологом на основании анамнеза, клинических данных, дополнительных обследований и генетического
- 29. Диагностика Подтвердить диагноз мышечной дистрофии Беккера позволяет консультация генетика с проведением анализа ДНК. Выявление дупликаций или
- 31. Скачать презентацию

Мышечная дистрофия Дюшенна.
-прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся
Мышечная дистрофия Дюшенна.
-прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся

Генетическое нарушение.
Патология появляется вследствие генной мутации, имеющей место в области хр21.
Генетическое нарушение.
Патология появляется вследствие генной мутации, имеющей место в области хр21.

Тип наследования.
Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи
Тип наследования.
Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи

Частота заболевания.
Распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков.
Частота заболевания.
Распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков.

Возраст манифестации.
Миодистрофия Дюшенна характеризуется началом в первые 1-5 лет жизни ребенка,
Возраст манифестации.
Миодистрофия Дюшенна характеризуется началом в первые 1-5 лет жизни ребенка,

Основные клинические проявления
Как правило, уже на 1-ом году жизни заметно некоторое
Основные клинические проявления
Как правило, уже на 1-ом году жизни заметно некоторое

Основные клинические проявления
Мышечная слабость возникает на 3-4-ом годах жизни.
Первоначально она
Основные клинические проявления
Мышечная слабость возникает на 3-4-ом годах жизни.
Первоначально она
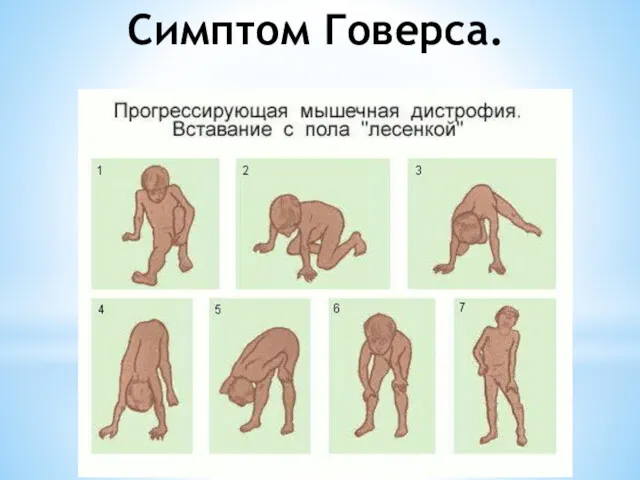
Симптом Говерса.
Симптом Говерса.
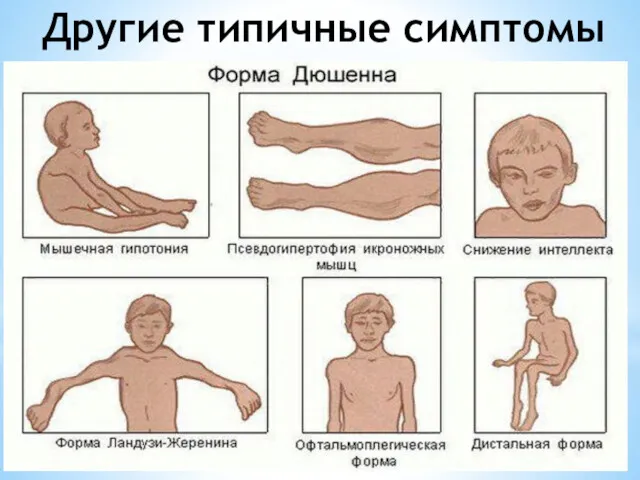
Другие типичные симптомы
Другие типичные симптомы

Основные клинические проявления
Мышечные атрофии начинаются с мышц бедер и тазового пояса.
Основные клинические проявления
Мышечные атрофии начинаются с мышц бедер и тазового пояса.

Основные клинические проявления
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе.
Основные клинические проявления
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе.

Основные клинические проявления.
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным
Основные клинические проявления.
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным

Диагностика
Установить диагноз миодистрофии Дюшенна помогают:
анамнез
неврологическое обследование
результаты электрофизиологического тестирования
Диагностика
Установить диагноз миодистрофии Дюшенна помогают:
анамнез
неврологическое обследование
результаты электрофизиологического тестирования

Диагностика
Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа,
Диагностика
Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа,

Диагностика
В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не
Диагностика
В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не

Специфическое лечение
Специфического лечения в настоящее время не существует. Лечение только симтоматическое:
Для
Специфическое лечение
Специфического лечения в настоящее время не существует. Лечение только симтоматическое:
Для

Справка.
В норме белок дистрофин поддерживает целостность сарколеммы - мембраны миоцитов (мышечных
Справка.
В норме белок дистрофин поддерживает целостность сарколеммы - мембраны миоцитов (мышечных

Мышечная дистрофия Беккера.
вариант наследственной сцепленной с Х-хромосомой миодистрофии, отличающейся более замедленным
Мышечная дистрофия Беккера.
вариант наследственной сцепленной с Х-хромосомой миодистрофии, отличающейся более замедленным

Генетическое нарушение
В основе заболевания лежит мутация в гене, ответственном за кодирование
Генетическое нарушение
В основе заболевания лежит мутация в гене, ответственном за кодирование

Тип наследования и частота заболевания.
Патология наследуется рецессивно сцеплено с Х-хромосомой, поэтому
Тип наследования и частота заболевания.
Патология наследуется рецессивно сцеплено с Х-хромосомой, поэтому

Возраст манифестации.
Прогрессирующая мышечная дистрофия Беккера манифестирует обычно в период от 10
Возраст манифестации.
Прогрессирующая мышечная дистрофия Беккера манифестирует обычно в период от 10

Основные клинические симптомы.
Начальными признаками заболевания выступают чрезмерная утомляемость и мышечная слабость
Основные клинические симптомы.
Начальными признаками заболевания выступают чрезмерная утомляемость и мышечная слабость

Основные клинические симптомы.
Как и другие наследственные миопатии, заболевание Беккера характеризуется симметрично
Основные клинические симптомы.
Как и другие наследственные миопатии, заболевание Беккера характеризуется симметрично

Основные клинические симптомы
Клиническая картина мышечной дистрофии Беккера во многом сходна с
Основные клинические симптомы
Клиническая картина мышечной дистрофии Беккера во многом сходна с
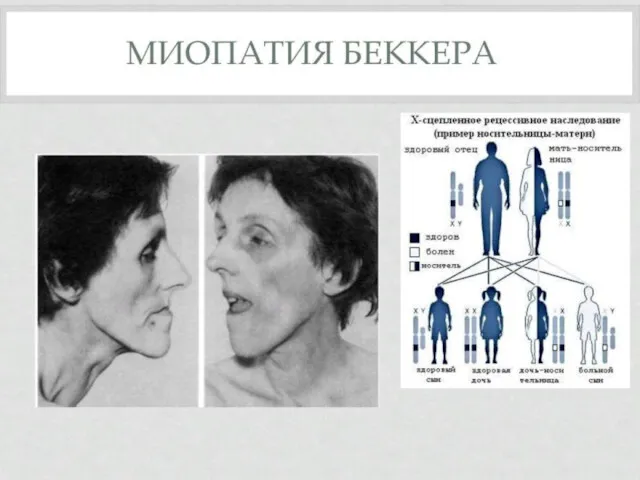
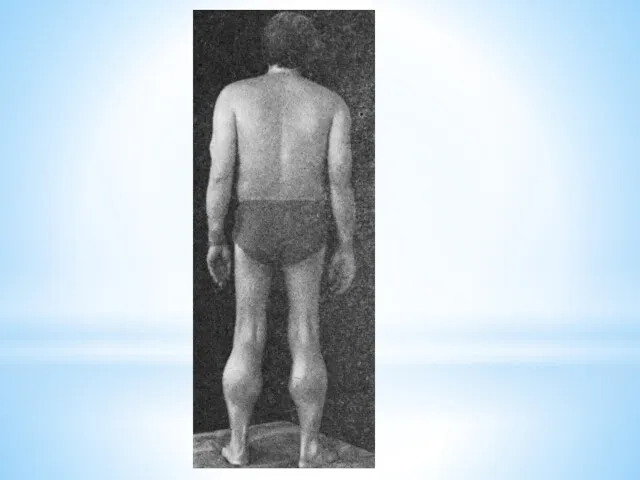

Диагностика.
Прогрессирующая мышечная дистрофия Беккера диагностируется неврологом на основании анамнеза, клинических данных,
Диагностика.
Прогрессирующая мышечная дистрофия Беккера диагностируется неврологом на основании анамнеза, клинических данных,

Диагностика
Подтвердить диагноз мышечной дистрофии Беккера позволяет консультация генетика с проведением анализа
Диагностика
Подтвердить диагноз мышечной дистрофии Беккера позволяет консультация генетика с проведением анализа
 Сердечно-сосудистые заболевания при ДТЗ
Сердечно-сосудистые заболевания при ДТЗ Загальна геріатрія. Особливості спостереження і догляду за хворими похилого та старечого віку
Загальна геріатрія. Особливості спостереження і догляду за хворими похилого та старечого віку Пластика деформаций лица лоскутами на ножке
Пластика деформаций лица лоскутами на ножке Антипсихотики (нейролептики)
Антипсихотики (нейролептики) Вирустар генетикасы. Вирустық геномның ұйымдасуы. Вирустық геномдардың репликациясы
Вирустар генетикасы. Вирустық геномның ұйымдасуы. Вирустық геномдардың репликациясы Инсулиновая помпа. Болюсное введение
Инсулиновая помпа. Болюсное введение Современные методы обследования больных с опухолями головы и шеи
Современные методы обследования больных с опухолями головы и шеи Прогноз и реабилитация больных в современной детской хирургии
Прогноз и реабилитация больных в современной детской хирургии Воспалительные заболевания позвоночника
Воспалительные заболевания позвоночника Заманауи эндодонттық аспаптар
Заманауи эндодонттық аспаптар Наследственные заболевания человека. Фенилкетонурия
Наследственные заболевания человека. Фенилкетонурия Острый аппендицит
Острый аппендицит Гипоксия
Гипоксия Введение в биологию
Введение в биологию Досье формата CTD
Досье формата CTD Система государственных учреждений, обеспечивающих контроль качества лекарственных средств
Система государственных учреждений, обеспечивающих контроль качества лекарственных средств Хирургические заболевания пищевода
Хирургические заболевания пищевода Зардап шеккендерге психологиялық көмек көрсету дағдыларын қалыптастыру
Зардап шеккендерге психологиялық көмек көрсету дағдыларын қалыптастыру Влияние биоритмов на проявление действия лекарственных средств. Понятие о хронофармакологии
Влияние биоритмов на проявление действия лекарственных средств. Понятие о хронофармакологии Жүйке жүйесі жұлын
Жүйке жүйесі жұлын Роль і значення лікарської етики і деонтології у загальній структурі соціального регулювання медичної діяльності
Роль і значення лікарської етики і деонтології у загальній структурі соціального регулювання медичної діяльності Категория Косметология
Категория Косметология Мониторинг и поддержание дыхания. Капнография
Мониторинг и поддержание дыхания. Капнография Наблюдение здорового ребенка на педиатрическом участке. Группы риска детей раннего возраста
Наблюдение здорового ребенка на педиатрическом участке. Группы риска детей раннего возраста Свойства материалов и их влияние на ткани зуба. Материаловедение. Лекция № 1. Тема 2
Свойства материалов и их влияние на ткани зуба. Материаловедение. Лекция № 1. Тема 2 Контроль работоспособности при занятиях футболом
Контроль работоспособности при занятиях футболом Cестринский уход за новорождёнными при многоплодной беременности
Cестринский уход за новорождёнными при многоплодной беременности Наиболее значимые для наркологической службы регламентирующие документы
Наиболее значимые для наркологической службы регламентирующие документы