- Спинальные амиотрофии

Содержание
- 2. Определение Наследственное прогрессирующее нервно-мышечное заболевание, основной механизм развития клинических признаков которого связан с прогрессирующей дегенерацией мотонейронов
- 3. Тип наследования: Аутосомно-рецессивный. Гены, ответственные за развитие заболевания: Ген SMN (survival motor neuron gene) расположен на
- 4. ЭТИОЛОГИЯ И ПАТОГЕНЕЗ Причины возникновения и механизмы развития наследственных амиотрофий изучены не до конца. В целом
- 5. Классификация 1. Спинальная амиотрофия тип I(болезнь Верднига- Гоффмана) 2. Спинальная амиотрофия тип II( болезнь Эмери-Дрейфиса) 3.
- 6. САМ I(болезнь Верднига- Гоффмана) Развивается в первые шесть месяцев жизни и характеризуется тяжелым злокачественным течением. Признаком
- 7. СAМ II( болезнь Эмери-Дрейфиса ) Характеризуется более поздним началом (от 6 до 18 месяцев) и менее
- 8. САМ III(болезнь Кугельберга-Веландер ) Возникает в широком возрастном диапазоне от 18 месяцев до 20 лет. Наиболее
- 9. САМ IV(взрослая форма) Медленно прогрессирующее заболевание, имеющее начало в большинстве случаев после 35 лет, значительно не
- 10. Дифференциальная диагностика Миопатии Острая форма полиомиелита Врожденные миотонии Атоническая форма ДЦП Наследственные патологии обмена веществ
- 11. Диагностика Важным является сбор данных о возможном наличии признаков заболевания у кого-то из семьи, что может
- 13. Скачать презентацию
Определение
Наследственное прогрессирующее нервно-мышечное заболевание, основной механизм развития клинических признаков которого связан
Определение
Наследственное прогрессирующее нервно-мышечное заболевание, основной механизм развития клинических признаков которого связан

Тип наследования:
Аутосомно-рецессивный.
Гены, ответственные за развитие заболевания:
Ген SMN (survival motor neuron gene) расположен на
Тип наследования:
Аутосомно-рецессивный.
Гены, ответственные за развитие заболевания:
Ген SMN (survival motor neuron gene) расположен на

ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Причины возникновения и механизмы развития наследственных амиотрофий изучены не
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Причины возникновения и механизмы развития наследственных амиотрофий изучены не

Классификация
1. Спинальная амиотрофия тип I(болезнь Верднига- Гоффмана)
2. Спинальная амиотрофия тип II( болезнь
Классификация
1. Спинальная амиотрофия тип I(болезнь Верднига- Гоффмана)
2. Спинальная амиотрофия тип II( болезнь

САМ I(болезнь Верднига- Гоффмана)
Развивается в первые шесть месяцев жизни и характеризуется
САМ I(болезнь Верднига- Гоффмана)
Развивается в первые шесть месяцев жизни и характеризуется

СAМ II( болезнь Эмери-Дрейфиса )
Характеризуется более поздним началом (от 6 до
СAМ II( болезнь Эмери-Дрейфиса )
Характеризуется более поздним началом (от 6 до
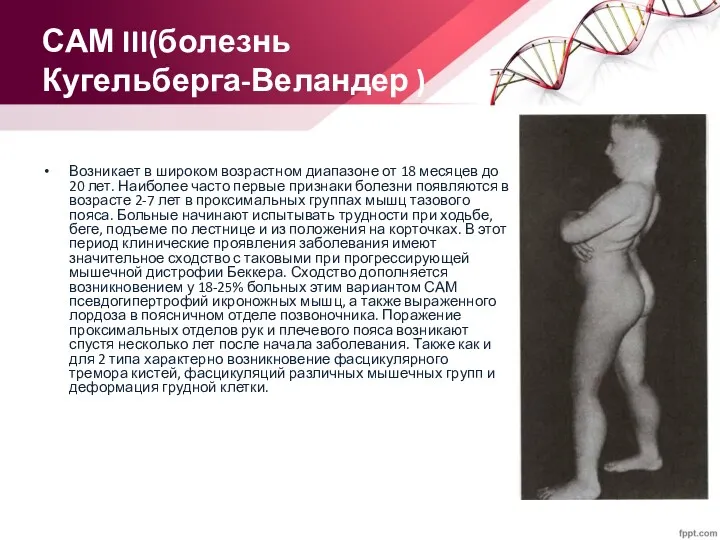
САМ III(болезнь Кугельберга-Веландер )
Возникает в широком возрастном диапазоне от 18 месяцев
САМ III(болезнь Кугельберга-Веландер )
Возникает в широком возрастном диапазоне от 18 месяцев

САМ IV(взрослая форма)
Медленно прогрессирующее заболевание, имеющее начало в большинстве случаев после
САМ IV(взрослая форма)
Медленно прогрессирующее заболевание, имеющее начало в большинстве случаев после

Дифференциальная
диагностика
Миопатии
Острая форма полиомиелита
Врожденные миотонии
Атоническая форма ДЦП
Наследственные патологии обмена веществ
Дифференциальная
диагностика
Миопатии
Острая форма полиомиелита
Врожденные миотонии
Атоническая форма ДЦП
Наследственные патологии обмена веществ

Диагностика
Важным является сбор данных о возможном наличии признаков заболевания у кого-то
Диагностика
Важным является сбор данных о возможном наличии признаков заболевания у кого-то
 Санатории Большой Ялты
Санатории Большой Ялты Иммунопатологические процессы. (Лекция 10)
Иммунопатологические процессы. (Лекция 10) ВИЧ-инфекция у наркопотребителей: что мы можем сделать сегодня?
ВИЧ-инфекция у наркопотребителей: что мы можем сделать сегодня? Антигены и антитела. (Лекция 2)
Антигены и антитела. (Лекция 2) Репродуктивті денсаулық
Репродуктивті денсаулық Дифференциальная диагностика болезней с желтухой
Дифференциальная диагностика болезней с желтухой Болезни желудка. Гастриты. Язвенная болезнь. Рак желудка
Болезни желудка. Гастриты. Язвенная болезнь. Рак желудка О коронавирусе детям
О коронавирусе детям Климактерический период
Климактерический период Прикормы, правила введения. Национальная программа оптимизации вскармливания детей первого года жизни в Российской Федерации
Прикормы, правила введения. Национальная программа оптимизации вскармливания детей первого года жизни в Российской Федерации Микробиология и иммунология. Классификация бактерий. Морфология бактерий
Микробиология и иммунология. Классификация бактерий. Морфология бактерий Лейкоцитозы, лейкопении
Лейкоцитозы, лейкопении Предмет епідеміології. Вчення про епідемічний процес. Напрями боротьби з інфекційними хворобами. Проблеми імунопрофілактики
Предмет епідеміології. Вчення про епідемічний процес. Напрями боротьби з інфекційними хворобами. Проблеми імунопрофілактики Лишний вес. Ожирение
Лишний вес. Ожирение Стоматология. Анализ клинического случая. Диагностика
Стоматология. Анализ клинического случая. Диагностика Компьютерная томография в диагностике повреждений органов живота и таза
Компьютерная томография в диагностике повреждений органов живота и таза Патология уха. Острый средний отит
Патология уха. Острый средний отит Этика в психиатрии и психотерапии
Этика в психиатрии и психотерапии Пародонт тінінің анатомо-топографиялық ерекшелігі
Пародонт тінінің анатомо-топографиялық ерекшелігі Қазақстандағы денсаулық сақтау ұйымы және бағдарламалары
Қазақстандағы денсаулық сақтау ұйымы және бағдарламалары Endocrine system
Endocrine system Жедел шашыранды склерозбен ауыратын ересек науқастарда никтурияны емдеу үшін мелотониннің тиімділігін анықтау
Жедел шашыранды склерозбен ауыратын ересек науқастарда никтурияны емдеу үшін мелотониннің тиімділігін анықтау Базидиальные грибы юга западной Сибири
Базидиальные грибы юга западной Сибири Инфекциялық әлеуметтік маңызы бар аурулар. Аса қауіпті инфекциялар (жұқпалар). Туберкулез
Инфекциялық әлеуметтік маңызы бар аурулар. Аса қауіпті инфекциялар (жұқпалар). Туберкулез Рак тела матки
Рак тела матки Геморрагический синдром. Острый лейкоз
Геморрагический синдром. Острый лейкоз Чума
Чума Острый панкеатит
Острый панкеатит