- Amyloidosis. Definitions

Содержание
- 2. Definitions Amyloidosis is a clinical disorder caused by extracellular deposition of insoluble abnormal fibrils that injure
- 3. Common features of all definitions presence of systemic protein metabolism disorder (acquired or hereditary) extracellular deposition
- 4. What is amyloid? (physical properties) straight, rigid, non-branching, of indeterminate length and 10 to 15nm in
- 5. What are β-pleated sheets single amyloid fibril consists of stacks of anti-parallel β-pleated sheets arranged with
- 6. Tertiary structure of amyloid proteins leading to insolubility
- 7. Amyloid fibers
- 8. In tissues
- 9. Chemical properties (main components) Proteins and their derivates Glucosaminoglycans amyloid P component Other proteins in amyloid
- 10. Main protein precursors (total 22) serum amyloid A protein (SAA) AL proteins (monoclonal light and heavy
- 11. Glycosaminoglycans significance in amyloid is unclear participate in organization of some normal structural proteins into fibrils;
- 12. amyloid P component and serum amyloid P component amyloid deposits in all different forms of the
- 13. Morphology and staining: common features for all types Amorphous eosinophilic appearance on light microscopy after hematoxylin
- 14. Typical staining for amyloid (right – heart Congo red, left – kidney Hematoxilin/eosin)
- 15. Classifications: before 1993 AA (inflammatory) AL (light chains related) AF (familial) AS (senile) AD (dermal) AH
- 16. WHO (1993): biochemical structure-based classification. Systemic variants AA (ApoSAA): chronic inflammatory diseases; periodical fever; Muckle-Wales AL
- 17. WHO (1993): local variants AL (Locally produced monoclonal Ig): local urogenital; skin, eyes, respiratory Aβ (β-amyloid
- 18. From 1993 to nowadays new precursors and new variants were found (2006 – 22 precursors). So,
- 19. Systemic Ig light chains (plasma cell disorders) Transthyretin (Familial amyloidosis, senile cardiac amyloidosis) A amyloidosis (Inflammation,
- 20. Hereditary (Familial systemic amyloidosis) Fibrinogen alpha chain Apolipoprotein AI Apolipoprotein AII Lysozyme
- 21. CNS amyloidosis Beta protein precursor (Alzheimer syndrome, Down syndrome, hereditary cerebral hemorrhage with amyloidosis - Dutch
- 22. Ocular Gelsolin (Familial amyloidosis; Finnish type) Lactoferrin (Familial corneal amyloidosis) Keratoepithelin (Familial corneal dystrophies)
- 23. Localized Calcitonin (Medullary thyroid carcinoma) IAAP= Amylin (Insulinoma, type 2 diabetes) Atrial natriuretic factor (Isolated atrial
- 24. Clinical syndromes related to amyloidosis General symptoms and intoxication: weakness, fatigue, sometimes fever and weight loss
- 25. Skin affection
- 26. Skin: papules on fingers
- 28. Skin: hemorrhages and papules
- 29. Skin microscopy
- 30. Periphreral nervous system: axonal peripheral neuropathy with subsequent demyelination: paresthesiae, numbness, muscular weakness; begin from lower
- 31. Central nervous system cerebral blood vessels affection recurrent cerebral hemorrhages intracerebral plaques progressive dementia
- 32. Gastrointestinal disorders: Tongue: increased, dense, red or purple; so that it can’t go in mouth; tooth
- 36. Heart affection
- 38. Heart: myocardium increase of relative cardiac dullness, soft heart sounds, systolic murmur at the apex and
- 39. ECG: heart muscle affection ECG – decrease of voltage, plain or inverted T, scars, pseudoinfarction QS
- 40. Heart: coronary arteries secondary coronary syndrome and myocardial infarction. more marked affection of intramyocardial arteries; angiographic
- 41. Rhythm and conductivity disorders conductivity disorders in sinus node, AV node and left bundle branch with
- 42. Pericardium and endocardium affection Pericardium deposits – constrictive pericarditis Valves affection (amyloid deposits in valves): mild
- 43. Echo The most common: thickening of the intraventricular septum (usually 15 mm and more; normal values
- 44. Thickening of the septum
- 45. Pericardial changes
- 48. ATTR
- 52. WHO staging system for cardiac amyloid 1 – no symptomatic or occult cardiac amyloid by biopsy
- 53. Vessels capillaries in the subcutaneous fat dermal capillars coronary and brain arteries (coronary syndrome, recurrent strokes)
- 54. Liver and spleen Hepatomegaly; usually elevation of alkaline phosphatase is revealed with near normal levels of
- 55. Kidneys
- 57. Kidneys: symptoms Proteinuria (usually – with nephrotic syndrome) Chronic renal failure Acute renal failure due to
- 58. Kidneys: staging system
- 59. Joints affection usually occurs in association with myeloma mimic acute polyarticular rheumatoid arthritis affecting large joints
- 60. Blood Acquired bleeding diathesis: - deficiency of factor X and sometimes factor IX, or increased fibrinolysis:
- 61. Respiratory system vocal cord infiltration associated with focal clonal immunocyte dyscrasia nodular or diffuse infiltrative form
- 62. tracheobronchial associated with focal clonal immunocyte dyscrasia nodular or diffuse infiltrative manifested by dyspnea, cough Occasionally
- 63. parenchymal nodular associated with focal clonal immunocyte dyscrasia solitary (amyloidoma) or multiple nodules in lung parenchyma;
- 64. diffuse alveolar septal usually is a manifestation of systemic AL amyloidosis associated with low grade monoclonal
- 65. intrathoracic lymphadenopathy usually manifestation of systemic AL amyloidosis (hilar or meduastinal amyloidosis) is uni- or bilateral
- 70. Eye visible or palpable periocular mass or tissue infiltration ptosis periocular discomfort or pain proptosis or
- 71. Endocrine and exocrine glands adrenal gland infiltration (hypoadrenalism) thyroid infiltration (hypothyroidism) IAAP – progressive loss of
- 72. Inflammatory amyloidosis Amyloid A (AA) most common form of systemic amyloidosis worldwide. characterized by extracellular tissue
- 73. SAA is an apolipoprotein of high density lipoprotein particles SAA is a major exquisitely sensitive acute
- 74. The circulating concentration: Normal - 3mg/l Rise to over 1000mg/l within 24 to 48h in ongoing
- 75. Pathogenesis Inflammation Macrophages activation: IL-1, 6 IL-1,6: hepatic transcription of the messenger RNA for SAA High
- 76. Causes chronic inflammatory disorders chronic local or systemic microbial infections malignant neoplasms and blood system diseases
- 77. Chronic inflammatory disorders Very often: rheumatoid arthritis and juvenile rheumatoid arthritis – in 10% of arthrites
- 78. Chronic local or systemic microbial infections tuberculosis and leprosy chronic osteomyelitis bronchiectasis chronic abscesses chronically infected
- 79. malignant neoplasms and blood system diseases The most frequent: diseases, causing fever, other systemic symptoms, and
- 80. Subcutaneous drug abuse AA amyloidosis was frequently observed among subcutaneous drug abusers in some cities in
- 81. Clinical symptoms Relating to the main disease General: weakness, weight loss Kidneys affection (up to renal
- 82. Course: Initially, disease is manifesting only by transient proteinuria, increasing in cases of main disease exacerbations.
- 83. Outcomes and complications: Main - chronic renal failure (end-stage - 5-10 years from 1st symptoms); the
- 84. Prognosis Depends on the course of the main disease Survival: 50% of patients die within 5
- 85. Familiar Mediterranean fever (recurrent polyserositis) and AA-amyloidosis Epidemiology: Incidence: in families with healthy parents: 18%; with
- 86. Morphology serosa: non-specific inflammation with hyperaemia and a cellular infiltrate synovia: pannus formation vascular changes -
- 87. Pathogenesis genetic nature immunological disturbances (higher incidence of autoimmune diseases and allergy in patients with Mediterrhanian
- 88. Clinical manifestations and syndromes 1. Onset: in childhood (1st decade of life – 50%; before 20
- 89. 2. Fever: may be even asymptomatic (afebrile mild attacks) abdominal pain attacks with fever up to
- 90. 3. Joints affection: from arthralgia to arthritis (24-84%, mean 55%) symptoms increase during the first 24-48h;
- 91. asymmetric, non-destructive mono- or oligoarthritis affecting the large joints; knees and ankles (small – rare) 1-2
- 92. 4. Chest (pleurisy) pain more than 50% pleural friction rub - rare small effusion in costophrenic
- 93. 5. Abdominal pain - almost in all patients attacks originate in one area, spread over whole
- 94. 6. Skin rash – 10-20% localization - extensor surfaces of legs; below knees, over ankle joints
- 95. 7. Other organs affection attacks of pericarditis (occasionally) severe headache during attacks transient ECG changes (myo-,
- 96. 8. Kidneys: AA-amyloidosis at the late stages the first sign is massive albuminuria; within several years
- 97. Amyloid deposits in other organs intestine adrenals heart ovaries pancreas muscles deposits are mostly perivascular.
- 98. Clinical variants with abdominal; thoracic, joint and fever syndromes dominating may vary in different life periods
- 99. Course first symptoms: sudden onset of asymptomatic fever, arthralgia, chest and abdominal pain. last for days
- 100. Factors influencing exacerbations physical exertion stress walking and standing pregnancy.
- 101. Outcomes end-stage chronic renal failure and death. adequate treatment can delay (but not stop) the disease
- 102. Immunoglobulin-related amyloidosis (AL) monoclonal plasma cell disorder, associated with gammapathies mostly related to light chains (AL-amyloidosis)
- 103. Conditions causing AL-amyloidosis Multiple myeloma Waldenstrom disease Monoclonal gammapathy of undetermined significance (MGUS)
- 104. Pathogenesis In L chains certain amino acid and glycosylation characteristics predispose to amyloid formation (why -
- 105. Epidemiology Incidence: annually, 1-5 cases per 100,000 people occur (may be higher basing on myeloma incidence
- 106. Symptoms Major systemic amyloidosis with affection of most organs described (except CNS) Most common initial symptoms:
- 107. Localized amyloid L-chain type most commonly in respiratory tract often remains localized may involve ureter or
- 108. Complications congestive heart failure, arrhythmias, or both (cause of death more than 50%) renal failure bleedings
- 109. Course and prognosis In the absence of chemotherapy always progressive course Rapid development of heart or
- 110. ATTR –amyloidosis TTR is a serum protein that transports thyroxine and retinol-binding protein. TTR monomer contains
- 111. Normal-sequence TTR senile cardiac amyloidosis (SCA). microscopic deposits are also found in many other organs -
- 112. Clinical manifestations; SSA in 25% of old patients clinically silent microscopic, systemic deposits of transthyretin (TTR)
- 113. Clinical manifestations; SCA may be silent or accompanied by significant impairment of cardiac function
- 114. TTR mutations accelerate the process of TTR amyloid formation mutations destabilize TTR monomers or tetramers and
- 115. Variants of TTR systemic familial amyloidosis FAP (family amyloid polyneuropathy) –Val30Met (Valin to Metionin in 30
- 116. Epidemiology Incidence: cardiac ATTR with normal sequence – 15% of all the autopsies after 80 years
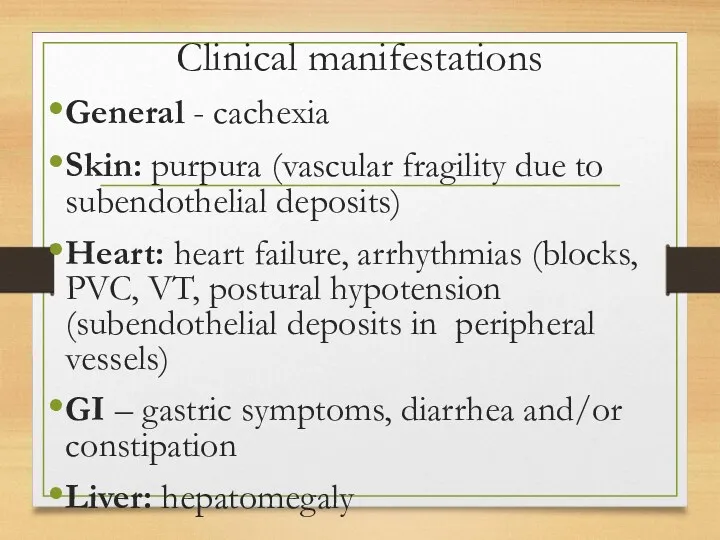
- 117. Clinical manifestations General - cachexia Skin: purpura (vascular fragility due to subendothelial deposits) Heart: heart failure,
- 118. Neuropathy: axonal degeneration of small nerve fibers due to deposits sensorimotor impairment (V30M - lower limb
- 119. FAP (family amyloid polyneuropathy) V30M major foci - Portugal, Japan, Sweden; age 20-70 Clinical manifestations include:
- 120. Beta2 –microglobulin (Dialysis-associated) Beta-2-microglobulin amyloidosis is a condition affecting patients on long-term hemodialysis or continuous ambulatory
- 121. Pathogenesis Beta-2-microglobulin is a component of beta chain of HLA class I molecule and is present
- 122. Epidemiology 1st symptoms – 4-8 years after haemodialysis onset (in 20%) 10 years after – in
- 123. Clinical manifestations 1. Neurological syndromes: carpal tunnel syndrome – most common (deposits in hands ligaments compress
- 124. Joints and bones affection Flexor tenosynovitis Scapulohumeral arthropathy - shoulder pain worse in supine position Spondyloarthropathy
- 125. Systemic manifestations after 10-15 years, usually asymptomatic GI: macroglossia, dysphagia, small bowel ischemia, malabsorption, and pseudoobstruction
- 126. Familial Renal (FRA) Syndrome of familial systemic amyloidosis with predominant nephropathy First described in 1932 by
- 127. Amyloid precursors Lysozyme Apolypoprotein I Apolipoprotein AII Fibrinogen A alpha-chain
- 128. Lysozyme Ile56Thr, Asp67His, Try64Arg Renal: Proteinuria and renal failure GI tract - Bleeding and perforation Liver
- 129. Apolypoprotein I Proteinuria and renal failure – almost in all Peptic ulcers (Gly26Arg ) Progressive neuropathy
- 130. Apolipoprotein AI with normal sequence Causes amyloid deposits in human aortic atherosclerotic plaques Found in 20-30%
- 131. Apolipoprotein AII Proteinuria and renal failure
- 132. Fibrinogen A alpha-chain Proteinuria and renal failure In Glu526Val variant hepatosplenomegaly and liver failure may occur
- 133. CNS amyloidosis Beta protein precursor (Alzheimer syndrome, Down syndrome, hereditary cerebral hemorrhage with amyloidosis - Dutch
- 134. Hereditary cerebral haemorrhage with amyloidosis; hereditary cerebral amyloid angiopathy Icelandic type autosomal dominant; symptoms early adult
- 135. Dutch type autosomal dominant; starts at middle age β-protein deposits recurrent normotensive cerebral hemorrhages Multi-infarct dementia;
- 136. Diagnosis of amyloidosis 1. Presence of amyloid: congo red staining 2. Type of amyloid: immunohistochemistry 3.
- 137. Tissues for biopsy subcutaneous fat aspiration (provides enough material for all investigations) – 60% rectal biopsy
- 139. AA SAA precursor level in blood Serum immunoglobulins (to exclude AL;in AA amyloidosis usually polyclonal hypergammaglobulinemia
- 140. Instrumental methods Avoid IV pyelography if amyloidosis is suspected (more frequent renal failure) Ultrasonography: kidneys’ size
- 142. AL Monoclonal immunoglobulin L chain - in the serum or the urine of 80-90% immunoglobulin free
- 143. Biochemistry concentration of normal Ig is often decreased hypogammaglobulinemia + proteinuria suggests a diagnosis of amyloid
- 144. Functional systems tests clotting system abnormalities kidney function tests liver function tests
- 145. Instrumental Echocardiography Radiolabeled pentagonal (P) component scanning: total body burden of amyloid Bone imaging: to reveal
- 146. ATTR subcutaneous fat aspiration sural nerve biopsy rectum, stomach, myocardium biopsy Congo red; antiserum against TTR
- 147. Instrumental Echocardiography Nerve conduction studies to monitor course of disease and assess response to treatment Genetic
- 148. Familial systemic (renal) Biopsy: amyloid confirmation Affection of organs SAP component scintigraphy; iodine I123 –labeled SAP
- 149. beta-2-microglobulin reference range of serum beta-2-microglobulin concentration of is 1.5-3 mg/L; can be elevated to values
- 150. Radiologic: joint erosions (usually large joints) lytic and cystic bone lesions (typically juxta-articular) pathological fractures spondyloarthropathies
- 151. CT amyloid deposits: intermediate attenuation. identification pseudotumors and pseudocystic areas in the juxta-articular bone. best method
- 152. MRI differentiating destructive spondyloarthropathies from inflammatory processes and infections.
- 153. Ultrasound tendon thickness. rotator cuff thickness greater than 8 mm, thickening of joint capsules (especially of
- 154. Scintigraphy radiolabeled P-component scans: iodine I 123 serum amyloid P iodohippurate sodium I 131 beta-2-microglobulin I
- 155. Biopsy with Congo red staining and with immunostaining centrifuged synovial fluid sediments cystic bone lesions biopsy
- 158. Treatment: AA primary inflammatory disease treatment tumor necrosis factor-a inhibitors and interleukin-1 inhibitors (arthritis, FMF) colchicine
- 161. melphalan plus prednisone melphalan, prednisone, and colchicine Other chemotherapeutic regimens used for multiple myeloma are also
- 162. Treatment of localized amyloid L-chain type has not been studied systematically chemotherapy is not indicated Localized
- 163. TTR Digoxin and calcium channel blockers are contraindicated Liver transplantation patients with cardiac, leptomeningeal, gastrointestinal, or
- 164. beta-2-microglobulin no adequate treatment (symptomatic) Improvement of dialysis membranes Online hemodiafiltration Direct hemoperfusion-type adsorption column (Lixelle):
- 166. Скачать презентацию

Definitions
Amyloidosis is a clinical disorder caused by extracellular deposition of insoluble
Definitions
Amyloidosis is a clinical disorder caused by extracellular deposition of insoluble

Common features of all definitions
presence of systemic protein metabolism disorder (acquired
Common features of all definitions
presence of systemic protein metabolism disorder (acquired

What is amyloid? (physical properties)
straight, rigid, non-branching, of indeterminate length and
What is amyloid? (physical properties)
straight, rigid, non-branching, of indeterminate length and

What are β-pleated sheets
single amyloid fibril consists
of stacks of anti-parallel
What are β-pleated sheets
single amyloid fibril consists
of stacks of anti-parallel
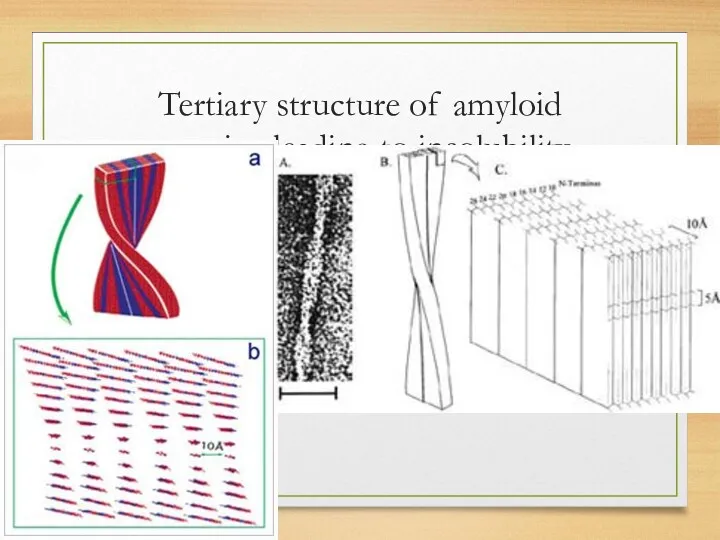
Tertiary structure of amyloid proteins leading to insolubility
Tertiary structure of amyloid proteins leading to insolubility
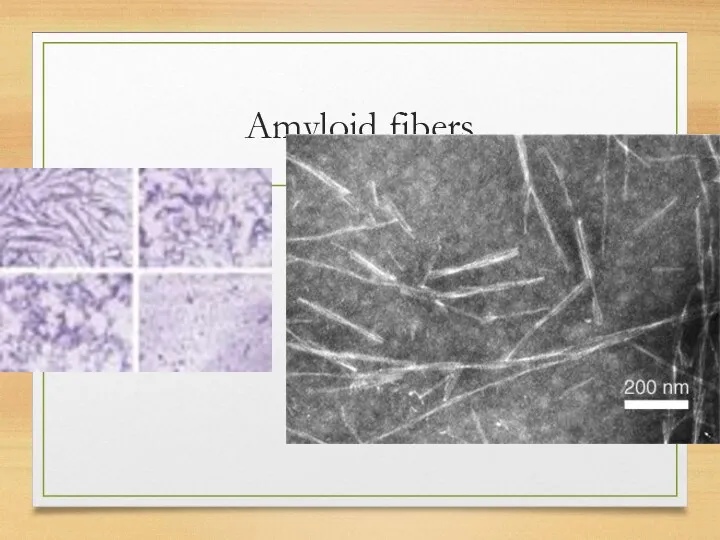
Amyloid fibers
Amyloid fibers
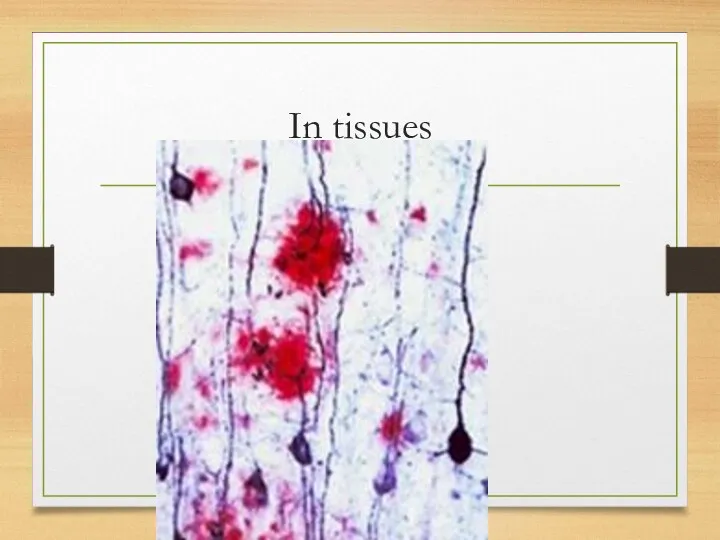
In tissues
In tissues

Chemical properties (main components)
Proteins and their derivates
Glucosaminoglycans
amyloid P component
Other proteins
Chemical properties (main components)
Proteins and their derivates
Glucosaminoglycans
amyloid P component
Other proteins

Main protein precursors (total 22)
serum amyloid A protein (SAA)
AL proteins
Main protein precursors (total 22)
serum amyloid A protein (SAA)
AL proteins

Glycosaminoglycans
significance in amyloid is unclear
participate in organization of some normal structural
Glycosaminoglycans
significance in amyloid is unclear
participate in organization of some normal structural

amyloid P component and serum amyloid P component
amyloid deposits in
amyloid P component and serum amyloid P component
amyloid deposits in

Morphology and staining: common features for all types
Amorphous eosinophilic appearance on
Morphology and staining: common features for all types
Amorphous eosinophilic appearance on
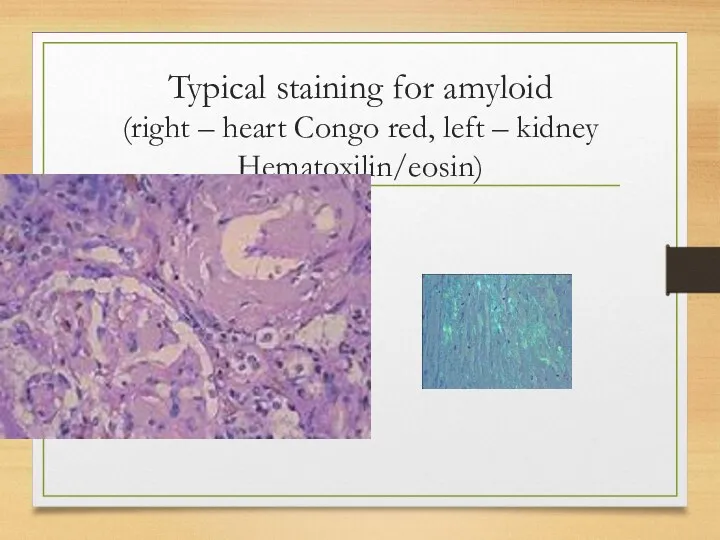
Typical staining for amyloid
(right – heart Congo red, left –
Typical staining for amyloid (right – heart Congo red, left –

Classifications: before 1993
AA (inflammatory)
AL (light chains related)
AF (familial)
AS (senile)
AD (dermal)
AH (haemodyalysis-related)
Classifications: before 1993
AA (inflammatory)
AL (light chains related)
AF (familial)
AS (senile)
AD (dermal)
AH (haemodyalysis-related)

WHO (1993): biochemical structure-based classification. Systemic variants
AA (ApoSAA): chronic inflammatory diseases;
WHO (1993): biochemical structure-based classification. Systemic variants
AA (ApoSAA): chronic inflammatory diseases;

WHO (1993): local variants
AL (Locally produced monoclonal Ig): local urogenital; skin,
WHO (1993): local variants
AL (Locally produced monoclonal Ig): local urogenital; skin,

From 1993 to nowadays new precursors and new variants were found
From 1993 to nowadays new precursors and new variants were found

Systemic
Ig light chains (plasma cell disorders)
Transthyretin (Familial amyloidosis, senile cardiac amyloidosis)
A
Systemic
Ig light chains (plasma cell disorders)
Transthyretin (Familial amyloidosis, senile cardiac amyloidosis)
A

Hereditary
(Familial systemic amyloidosis)
Fibrinogen alpha chain
Apolipoprotein AI
Apolipoprotein AII
Lysozyme
Hereditary
(Familial systemic amyloidosis)
Fibrinogen alpha chain
Apolipoprotein AI
Apolipoprotein AII
Lysozyme

CNS amyloidosis
Beta protein precursor (Alzheimer syndrome, Down syndrome, hereditary cerebral hemorrhage
CNS amyloidosis
Beta protein precursor (Alzheimer syndrome, Down syndrome, hereditary cerebral hemorrhage

Ocular
Gelsolin (Familial amyloidosis; Finnish type)
Lactoferrin (Familial corneal amyloidosis)
Keratoepithelin (Familial corneal
Ocular
Gelsolin (Familial amyloidosis; Finnish type)
Lactoferrin (Familial corneal amyloidosis)
Keratoepithelin (Familial corneal
Localized
Calcitonin (Medullary thyroid carcinoma)
IAAP= Amylin (Insulinoma, type 2 diabetes)
Atrial natriuretic
Localized
Calcitonin (Medullary thyroid carcinoma)
IAAP= Amylin (Insulinoma, type 2 diabetes)
Atrial natriuretic

Clinical syndromes related to amyloidosis
General symptoms and intoxication: weakness, fatigue, sometimes
Clinical syndromes related to amyloidosis
General symptoms and intoxication: weakness, fatigue, sometimes
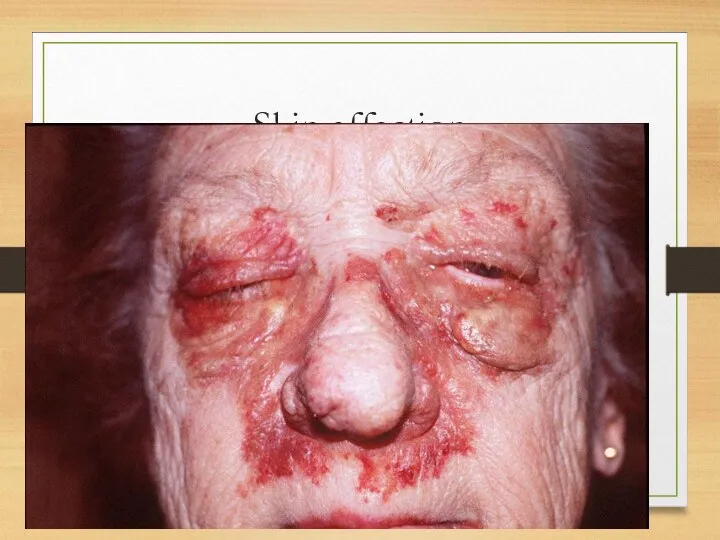
Skin affection
Skin affection
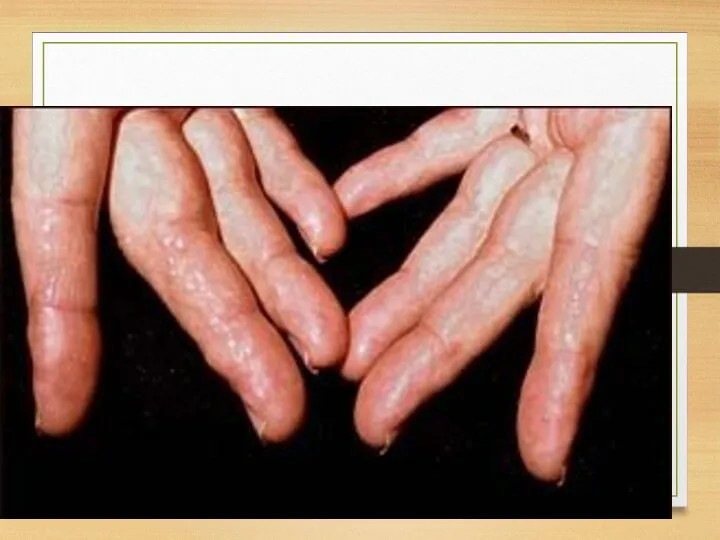
Skin: papules on fingers
Skin: papules on fingers
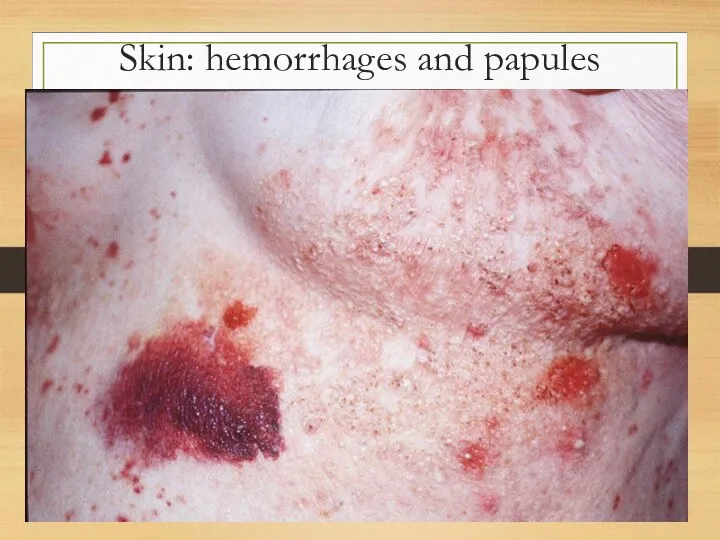
Skin: hemorrhages and papules
Skin: hemorrhages and papules
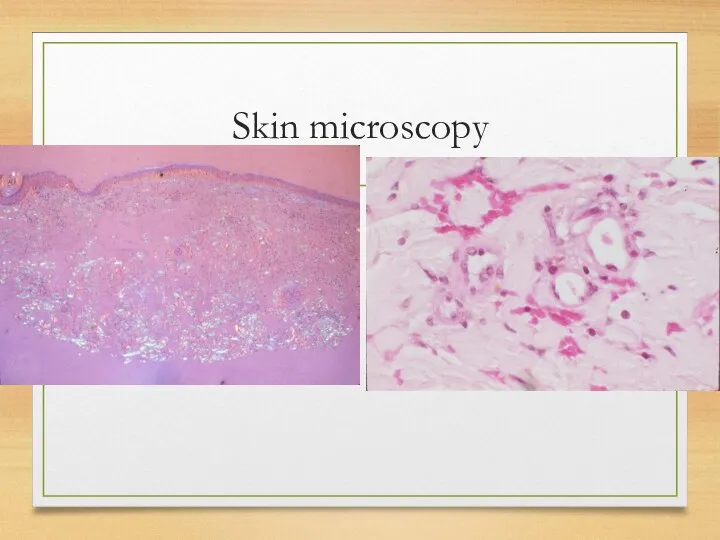
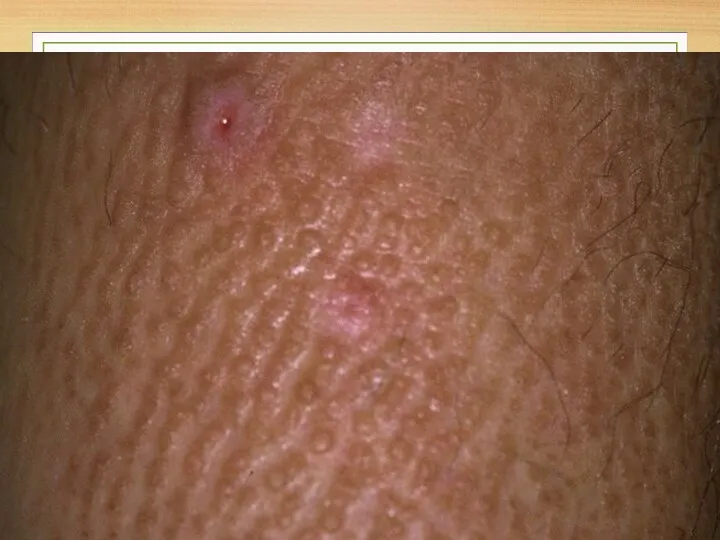
Skin microscopy
Skin microscopy
Periphreral nervous system:
axonal peripheral neuropathy with subsequent demyelination:
paresthesiae, numbness, muscular weakness;
Periphreral nervous system:
axonal peripheral neuropathy with subsequent demyelination:
paresthesiae, numbness, muscular weakness;

Central nervous system
cerebral blood vessels affection
recurrent cerebral hemorrhages
intracerebral plaques
progressive dementia
Central nervous system
cerebral blood vessels affection
recurrent cerebral hemorrhages
intracerebral plaques
progressive dementia

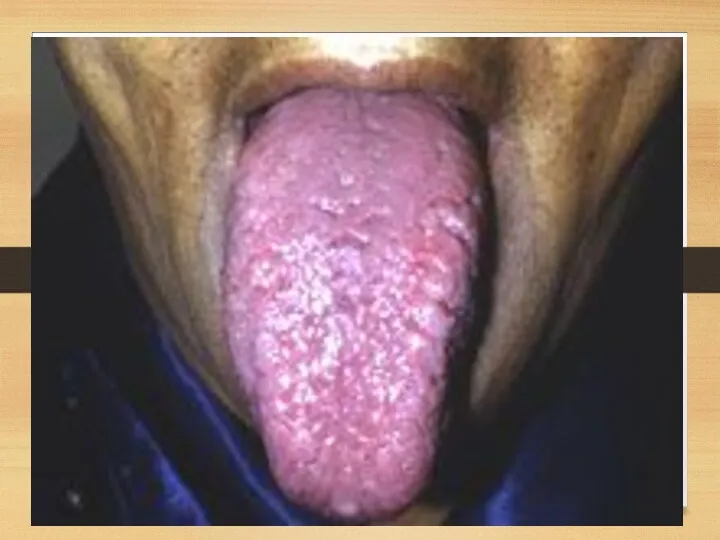
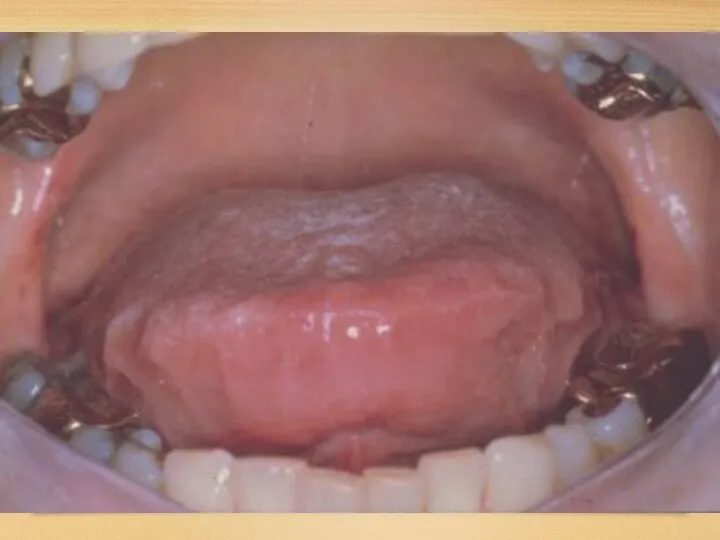
Gastrointestinal disorders:
Tongue: increased, dense, red or purple; so that it can’t
Gastrointestinal disorders:
Tongue: increased, dense, red or purple; so that it can’t
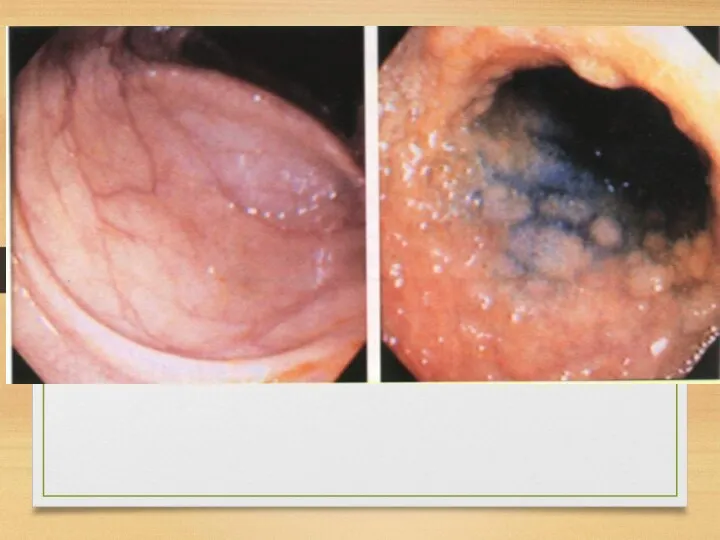
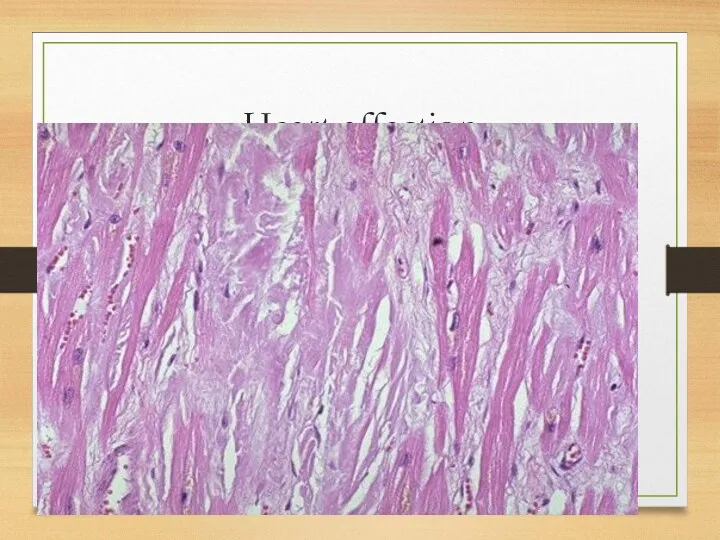
Heart affection
Heart affection
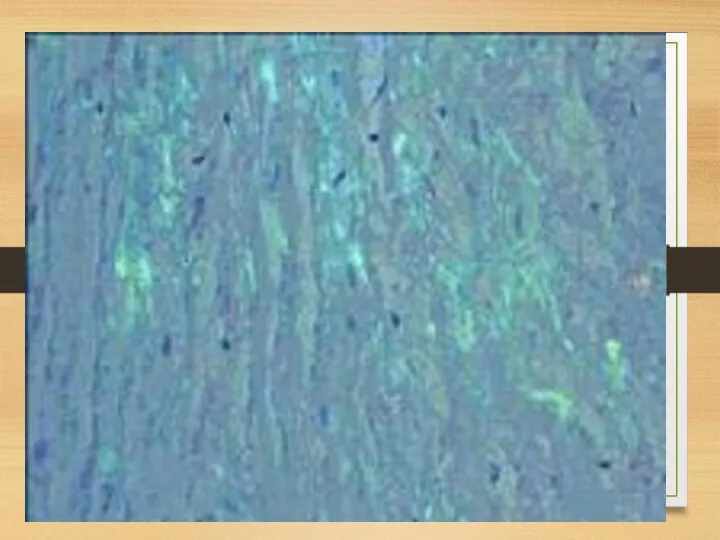

Heart: myocardium
increase of relative cardiac dullness, soft heart sounds, systolic
Heart: myocardium
increase of relative cardiac dullness, soft heart sounds, systolic

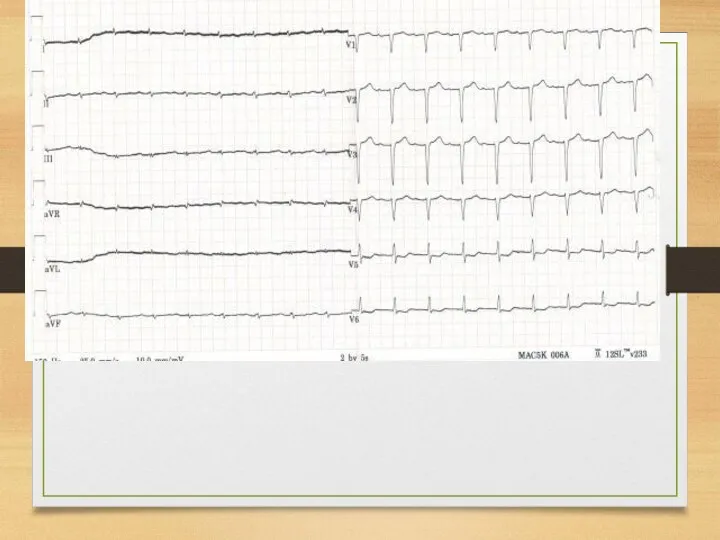
ECG: heart muscle affection
ECG – decrease of voltage, plain or inverted
ECG: heart muscle affection
ECG – decrease of voltage, plain or inverted

Heart: coronary arteries
secondary coronary syndrome and myocardial infarction.
more marked affection
Heart: coronary arteries
secondary coronary syndrome and myocardial infarction.
more marked affection

Rhythm and conductivity disorders
conductivity disorders in sinus node, AV node and
Rhythm and conductivity disorders
conductivity disorders in sinus node, AV node and

Pericardium and endocardium affection
Pericardium deposits – constrictive pericarditis
Valves affection (amyloid
Pericardium and endocardium affection
Pericardium deposits – constrictive pericarditis
Valves affection (amyloid

Echo
The most common: thickening of the intraventricular septum (usually 15 mm
Echo
The most common: thickening of the intraventricular septum (usually 15 mm
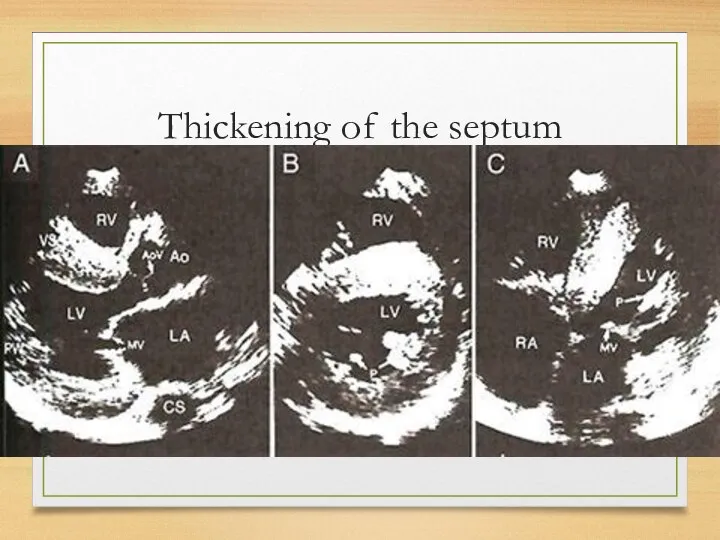
Thickening of the septum
Thickening of the septum
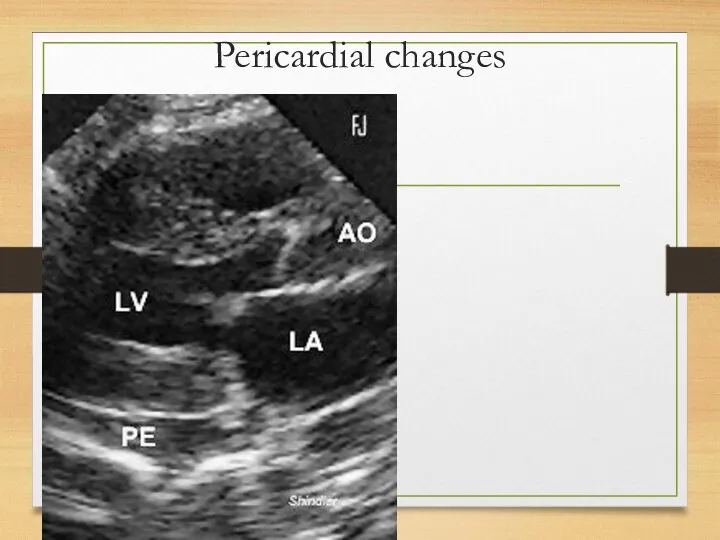
Pericardial changes
Pericardial changes
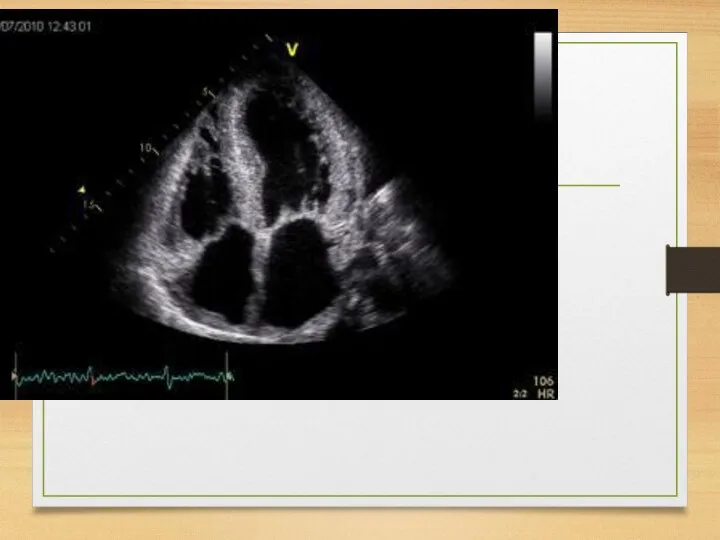
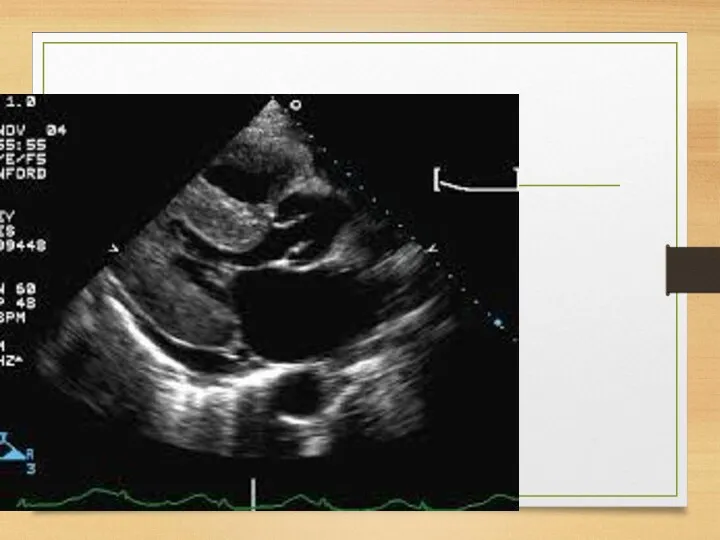
ATTR
ATTR
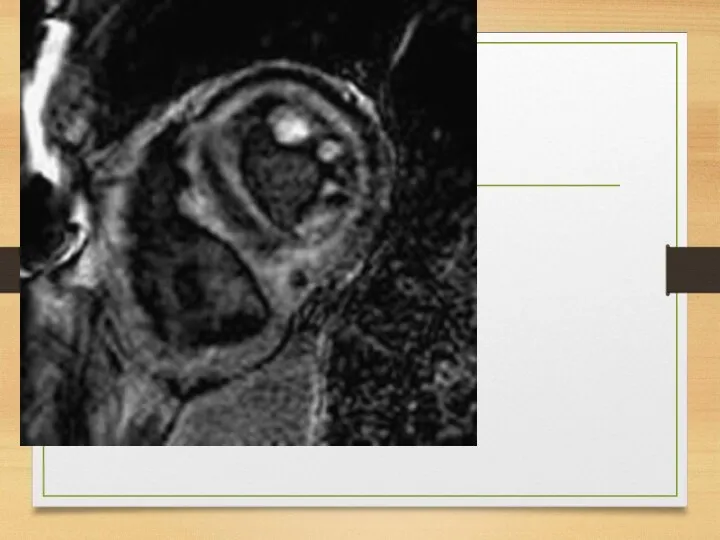
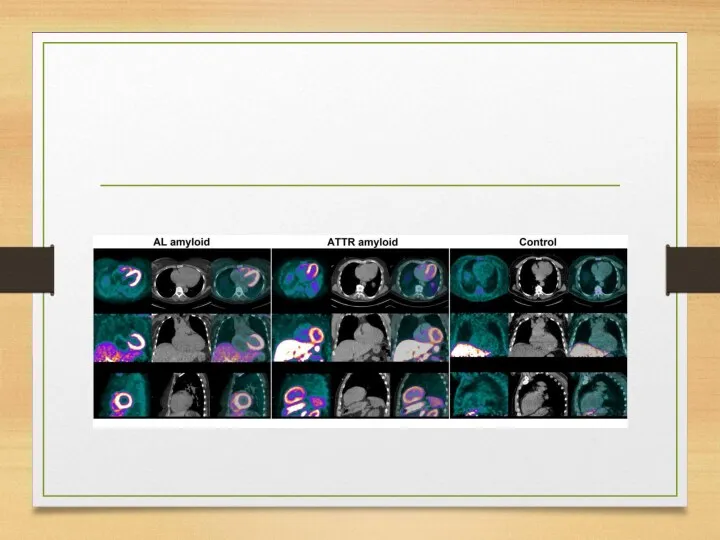
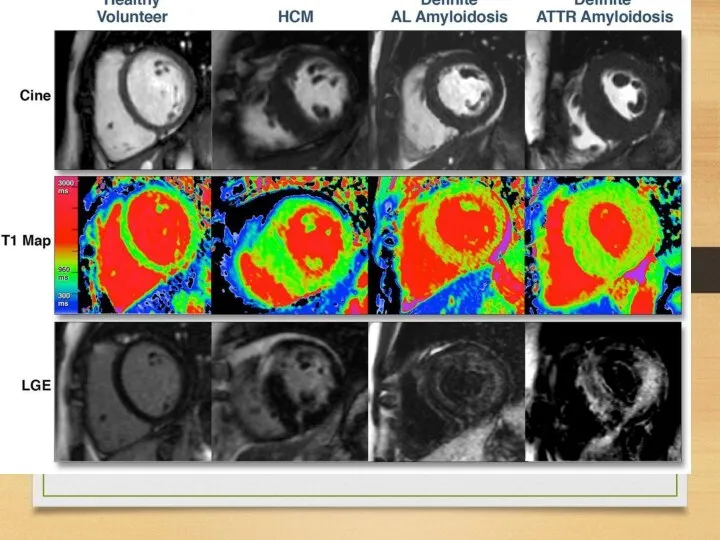

WHO staging system for cardiac amyloid
1 – no symptomatic or occult
WHO staging system for cardiac amyloid
1 – no symptomatic or occult

Vessels
capillaries in the subcutaneous fat
dermal capillars
coronary and brain arteries (coronary syndrome,
Vessels
capillaries in the subcutaneous fat
dermal capillars
coronary and brain arteries (coronary syndrome,

Liver and spleen
Hepatomegaly; usually elevation of alkaline phosphatase is revealed
Liver and spleen
Hepatomegaly; usually elevation of alkaline phosphatase is revealed
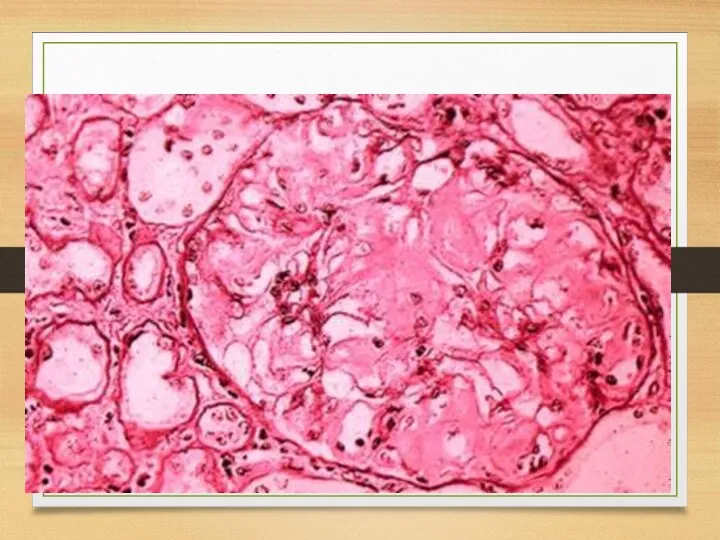
Kidneys
Kidneys
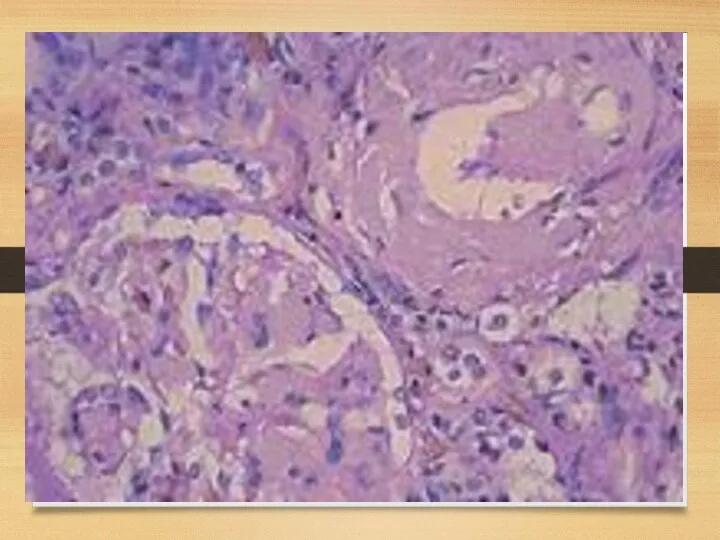

Kidneys: symptoms
Proteinuria (usually – with nephrotic syndrome)
Chronic renal failure
Acute renal failure
Kidneys: symptoms
Proteinuria (usually – with nephrotic syndrome)
Chronic renal failure
Acute renal failure
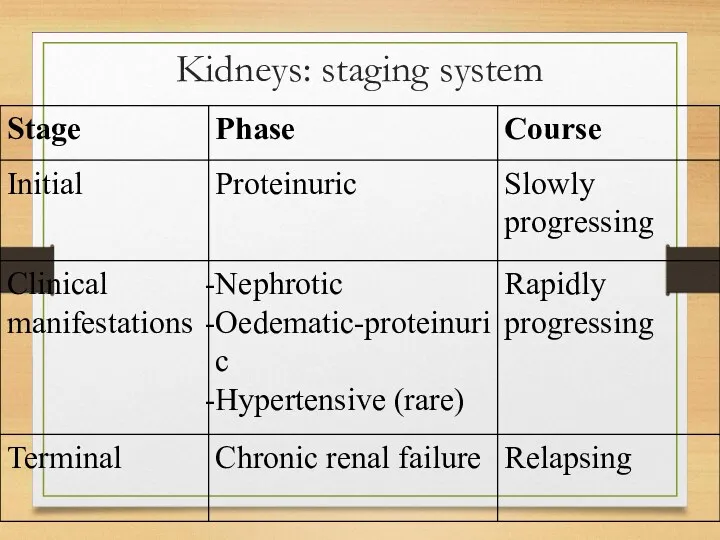
Kidneys: staging system
Kidneys: staging system

Joints affection
usually occurs in association with myeloma
mimic acute polyarticular
Joints affection
usually occurs in association with myeloma
mimic acute polyarticular

Blood
Acquired bleeding diathesis:
- deficiency of factor X and sometimes factor IX,
Blood
Acquired bleeding diathesis:
- deficiency of factor X and sometimes factor IX,

Respiratory system
vocal cord infiltration
associated with focal clonal immunocyte dyscrasia
nodular or
Respiratory system
vocal cord infiltration
associated with focal clonal immunocyte dyscrasia
nodular or

tracheobronchial
associated with focal clonal immunocyte dyscrasia
nodular or diffuse infiltrative
manifested by
tracheobronchial
associated with focal clonal immunocyte dyscrasia
nodular or diffuse infiltrative
manifested by

parenchymal nodular
associated with focal clonal immunocyte dyscrasia
solitary (amyloidoma) or multiple
parenchymal nodular
associated with focal clonal immunocyte dyscrasia
solitary (amyloidoma) or multiple

diffuse alveolar septal
usually is a manifestation of systemic AL
diffuse alveolar septal
usually is a manifestation of systemic AL

intrathoracic lymphadenopathy
usually manifestation of systemic AL amyloidosis (hilar or meduastinal amyloidosis)
is
intrathoracic lymphadenopathy
usually manifestation of systemic AL amyloidosis (hilar or meduastinal amyloidosis)
is
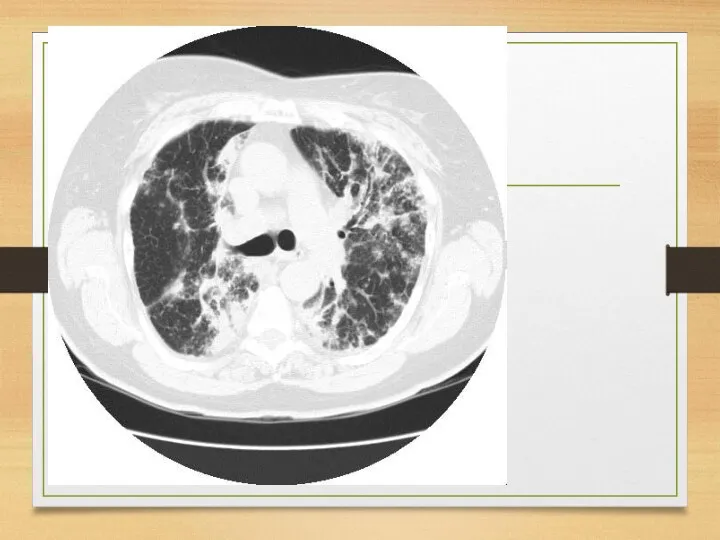
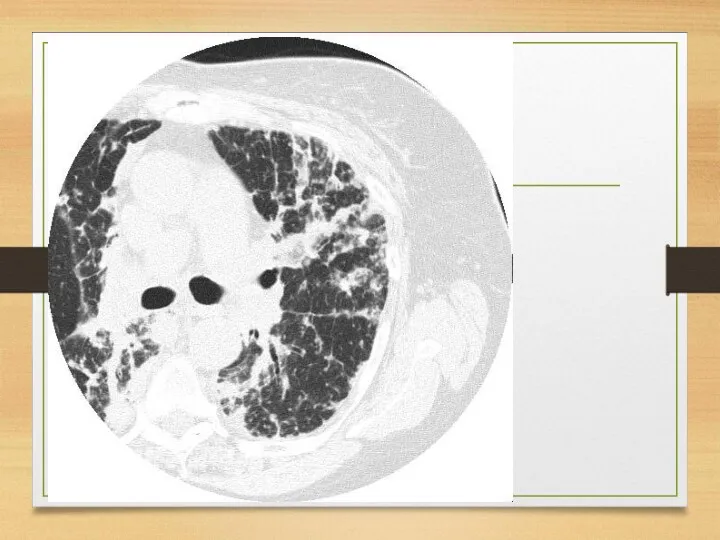
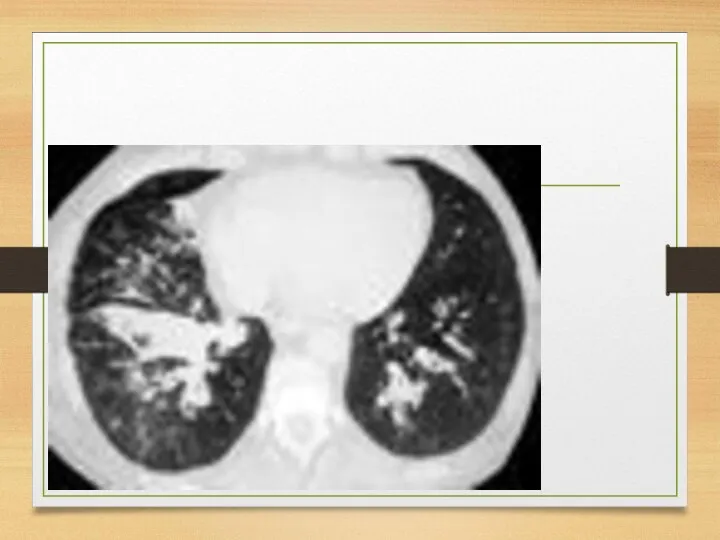

Eye
visible or palpable periocular mass or tissue infiltration
ptosis
periocular discomfort
Eye
visible or palpable periocular mass or tissue infiltration
ptosis
periocular discomfort

Endocrine and exocrine glands
adrenal gland infiltration (hypoadrenalism)
thyroid infiltration (hypothyroidism)
IAAP – progressive
Endocrine and exocrine glands
adrenal gland infiltration (hypoadrenalism)
thyroid infiltration (hypothyroidism)
IAAP – progressive

Inflammatory amyloidosis
Amyloid A (AA)
most common form of systemic amyloidosis worldwide.
characterized
Inflammatory amyloidosis
Amyloid A (AA)
most common form of systemic amyloidosis worldwide.
characterized

SAA is an apolipoprotein of high density lipoprotein particles
SAA is a
SAA is an apolipoprotein of high density lipoprotein particles
SAA is a

The circulating concentration:
Normal - 3mg/l
Rise to over 1000mg/l within
The circulating concentration:
Normal - 3mg/l
Rise to over 1000mg/l within

Pathogenesis
Inflammation
Macrophages activation: IL-1, 6
IL-1,6:
hepatic transcription of the messenger RNA for
Pathogenesis
Inflammation
Macrophages activation: IL-1, 6
IL-1,6:
hepatic transcription of the messenger RNA for

Causes
chronic inflammatory disorders
chronic local or systemic microbial infections
malignant neoplasms and
Causes
chronic inflammatory disorders
chronic local or systemic microbial infections
malignant neoplasms and

Chronic inflammatory disorders
Very often:
rheumatoid arthritis and juvenile rheumatoid arthritis – in
Chronic inflammatory disorders
Very often:
rheumatoid arthritis and juvenile rheumatoid arthritis – in

Chronic local or systemic microbial infections
tuberculosis and leprosy
chronic osteomyelitis
bronchiectasis
chronic
Chronic local or systemic microbial infections
tuberculosis and leprosy
chronic osteomyelitis
bronchiectasis
chronic

malignant neoplasms and blood system diseases
The most frequent:
diseases, causing fever, other
malignant neoplasms and blood system diseases
The most frequent:
diseases, causing fever, other

Subcutaneous drug abuse
AA amyloidosis was frequently observed among subcutaneous drug abusers
Subcutaneous drug abuse
AA amyloidosis was frequently observed among subcutaneous drug abusers

Clinical symptoms
Relating to the main disease
General: weakness, weight loss
Kidneys
Clinical symptoms
Relating to the main disease
General: weakness, weight loss
Kidneys

Course:
Initially, disease is manifesting only by transient proteinuria, increasing in cases
Course:
Initially, disease is manifesting only by transient proteinuria, increasing in cases

Outcomes and complications:
Main - chronic renal failure (end-stage - 5-10 years
Outcomes and complications:
Main - chronic renal failure (end-stage - 5-10 years

Prognosis
Depends on the course of the main disease
Survival: 50% of patients
Prognosis
Depends on the course of the main disease
Survival: 50% of patients

Familiar Mediterranean fever (recurrent polyserositis)
and AA-amyloidosis
Epidemiology:
Incidence: in families with healthy
Familiar Mediterranean fever (recurrent polyserositis)
and AA-amyloidosis
Epidemiology:
Incidence: in families with healthy

Morphology
serosa: non-specific inflammation with hyperaemia and a cellular infiltrate
synovia: pannus
Morphology
serosa: non-specific inflammation with hyperaemia and a cellular infiltrate
synovia: pannus

Pathogenesis
genetic nature
immunological disturbances (higher incidence of autoimmune diseases and
Pathogenesis
genetic nature
immunological disturbances (higher incidence of autoimmune diseases and

Clinical manifestations and syndromes
1. Onset: in childhood (1st decade of life
Clinical manifestations and syndromes
1. Onset: in childhood (1st decade of life

2. Fever:
may be even asymptomatic (afebrile mild attacks)
abdominal pain attacks
2. Fever:
may be even asymptomatic (afebrile mild attacks)
abdominal pain attacks

3. Joints affection:
from arthralgia to arthritis (24-84%, mean 55%)
symptoms increase during
3. Joints affection:
from arthralgia to arthritis (24-84%, mean 55%)
symptoms increase during

asymmetric, non-destructive mono- or oligoarthritis affecting the large joints; knees and
asymmetric, non-destructive mono- or oligoarthritis affecting the large joints; knees and

4. Chest (pleurisy) pain
more than 50%
pleural friction rub - rare
small
4. Chest (pleurisy) pain
more than 50%
pleural friction rub - rare
small

5. Abdominal pain - almost in all patients
attacks originate in one
5. Abdominal pain - almost in all patients
attacks originate in one

6. Skin rash – 10-20%
localization - extensor surfaces of legs; below
6. Skin rash – 10-20%
localization - extensor surfaces of legs; below

7. Other organs affection
attacks of pericarditis (occasionally)
severe headache during attacks
transient
7. Other organs affection
attacks of pericarditis (occasionally)
severe headache during attacks
transient

8. Kidneys: AA-amyloidosis
at the late stages
the first sign is massive albuminuria;
8. Kidneys: AA-amyloidosis
at the late stages
the first sign is massive albuminuria;

Amyloid deposits in other organs
intestine
adrenals
heart
ovaries
pancreas
muscles
deposits are mostly perivascular.
Amyloid deposits in other organs
intestine
adrenals
heart
ovaries
pancreas
muscles
deposits are mostly perivascular.

Clinical variants
with abdominal; thoracic, joint and fever syndromes dominating
may vary
Clinical variants
with abdominal; thoracic, joint and fever syndromes dominating
may vary

Course
first symptoms: sudden onset of asymptomatic fever, arthralgia, chest and abdominal
Course
first symptoms: sudden onset of asymptomatic fever, arthralgia, chest and abdominal

Factors influencing exacerbations
physical exertion
stress
walking and standing
pregnancy.
Factors influencing exacerbations
physical exertion
stress
walking and standing
pregnancy.

Outcomes
end-stage chronic renal failure and death.
adequate treatment can delay
Outcomes
end-stage chronic renal failure and death.
adequate treatment can delay

Immunoglobulin-related amyloidosis (AL)
monoclonal plasma cell disorder, associated with gammapathies
mostly related to
Immunoglobulin-related amyloidosis (AL)
monoclonal plasma cell disorder, associated with gammapathies
mostly related to

Conditions causing AL-amyloidosis
Multiple myeloma
Waldenstrom disease
Monoclonal gammapathy of undetermined significance (MGUS)
Conditions causing AL-amyloidosis
Multiple myeloma
Waldenstrom disease
Monoclonal gammapathy of undetermined significance (MGUS)

Pathogenesis
In L chains certain amino acid and glycosylation characteristics predispose to
Pathogenesis
In L chains certain amino acid and glycosylation characteristics predispose to

Epidemiology
Incidence: annually, 1-5 cases per 100,000 people occur (may be higher
Epidemiology
Incidence: annually, 1-5 cases per 100,000 people occur (may be higher

Symptoms
Major systemic amyloidosis with affection of most organs described (except CNS)
Most
Symptoms
Major systemic amyloidosis with affection of most organs described (except CNS)
Most

Localized amyloid L-chain type
most commonly in respiratory tract
often remains
Localized amyloid L-chain type
most commonly in respiratory tract
often remains

Complications
congestive heart failure, arrhythmias, or both (cause of death more than
Complications
congestive heart failure, arrhythmias, or both (cause of death more than

Course and prognosis
In the absence of chemotherapy always progressive course
Rapid
Course and prognosis
In the absence of chemotherapy always progressive course
Rapid

ATTR –amyloidosis
TTR is a serum protein that transports thyroxine and
ATTR –amyloidosis
TTR is a serum protein that transports thyroxine and

Normal-sequence TTR
senile cardiac amyloidosis (SCA).
microscopic deposits are also found in
Normal-sequence TTR
senile cardiac amyloidosis (SCA).
microscopic deposits are also found in
Clinical manifestations; SSA
in 25% of old patients clinically silent microscopic, systemic
Clinical manifestations; SSA
in 25% of old patients clinically silent microscopic, systemic

Clinical manifestations; SCA
may be silent or accompanied by significant impairment of
Clinical manifestations; SCA
may be silent or accompanied by significant impairment of

TTR mutations
accelerate the process of TTR amyloid formation
mutations destabilize
TTR mutations
accelerate the process of TTR amyloid formation
mutations destabilize

Variants of TTR systemic familial amyloidosis
FAP (family amyloid polyneuropathy) –Val30Met (Valin
Variants of TTR systemic familial amyloidosis
FAP (family amyloid polyneuropathy) –Val30Met (Valin

Epidemiology
Incidence:
cardiac ATTR with normal sequence – 15% of all the autopsies
Epidemiology
Incidence:
cardiac ATTR with normal sequence – 15% of all the autopsies
Clinical manifestations
General - cachexia
Skin: purpura (vascular fragility due to subendothelial
Clinical manifestations
General - cachexia
Skin: purpura (vascular fragility due to subendothelial

Neuropathy: axonal degeneration of small nerve fibers due to deposits
sensorimotor impairment
Neuropathy: axonal degeneration of small nerve fibers due to deposits
sensorimotor impairment

FAP (family amyloid polyneuropathy) V30M
major foci - Portugal, Japan, Sweden; age
FAP (family amyloid polyneuropathy) V30M
major foci - Portugal, Japan, Sweden; age

Beta2 –microglobulin (Dialysis-associated)
Beta-2-microglobulin amyloidosis is a condition affecting patients on
Beta2 –microglobulin (Dialysis-associated)
Beta-2-microglobulin amyloidosis is a condition affecting patients on

Pathogenesis
Beta-2-microglobulin is a component of beta chain of HLA class I
Pathogenesis
Beta-2-microglobulin is a component of beta chain of HLA class I

Epidemiology
1st symptoms – 4-8 years after haemodialysis onset (in 20%)
10 years
Epidemiology
1st symptoms – 4-8 years after haemodialysis onset (in 20%)
10 years

Clinical manifestations
1. Neurological syndromes:
carpal tunnel syndrome – most common (deposits in
Clinical manifestations
1. Neurological syndromes:
carpal tunnel syndrome – most common (deposits in

Joints and bones affection
Flexor tenosynovitis
Scapulohumeral arthropathy - shoulder pain worse in
Joints and bones affection
Flexor tenosynovitis
Scapulohumeral arthropathy - shoulder pain worse in

Systemic manifestations
after 10-15 years, usually asymptomatic
GI: macroglossia, dysphagia, small
Systemic manifestations
after 10-15 years, usually asymptomatic
GI: macroglossia, dysphagia, small

Familial Renal (FRA)
Syndrome of familial systemic amyloidosis with predominant nephropathy
First described
Familial Renal (FRA)
Syndrome of familial systemic amyloidosis with predominant nephropathy
First described

Amyloid precursors
Lysozyme
Apolypoprotein I
Apolipoprotein AII
Fibrinogen A alpha-chain
Amyloid precursors
Lysozyme
Apolypoprotein I
Apolipoprotein AII
Fibrinogen A alpha-chain

Lysozyme Ile56Thr, Asp67His, Try64Arg
Renal: Proteinuria and renal failure
GI tract - Bleeding
Lysozyme Ile56Thr, Asp67His, Try64Arg
Renal: Proteinuria and renal failure
GI tract - Bleeding

Apolypoprotein I
Proteinuria and renal failure – almost in all
Peptic ulcers (Gly26Arg
Apolypoprotein I
Proteinuria and renal failure – almost in all
Peptic ulcers (Gly26Arg

Apolipoprotein AI with normal sequence
Causes amyloid deposits in human aortic atherosclerotic
Apolipoprotein AI with normal sequence
Causes amyloid deposits in human aortic atherosclerotic

Apolipoprotein AII
Proteinuria and renal failure
Apolipoprotein AII
Proteinuria and renal failure

Fibrinogen A alpha-chain
Proteinuria and renal failure
In Glu526Val variant hepatosplenomegaly
Fibrinogen A alpha-chain
Proteinuria and renal failure
In Glu526Val variant hepatosplenomegaly

CNS amyloidosis
Beta protein precursor (Alzheimer syndrome, Down syndrome, hereditary cerebral hemorrhage
CNS amyloidosis
Beta protein precursor (Alzheimer syndrome, Down syndrome, hereditary cerebral hemorrhage

Hereditary cerebral haemorrhage with amyloidosis; hereditary cerebral amyloid angiopathy
Icelandic type
autosomal dominant;
Hereditary cerebral haemorrhage with amyloidosis; hereditary cerebral amyloid angiopathy
Icelandic type
autosomal dominant;

Dutch type
autosomal dominant; starts at middle age
β-protein deposits
recurrent
Dutch type
autosomal dominant; starts at middle age
β-protein deposits
recurrent

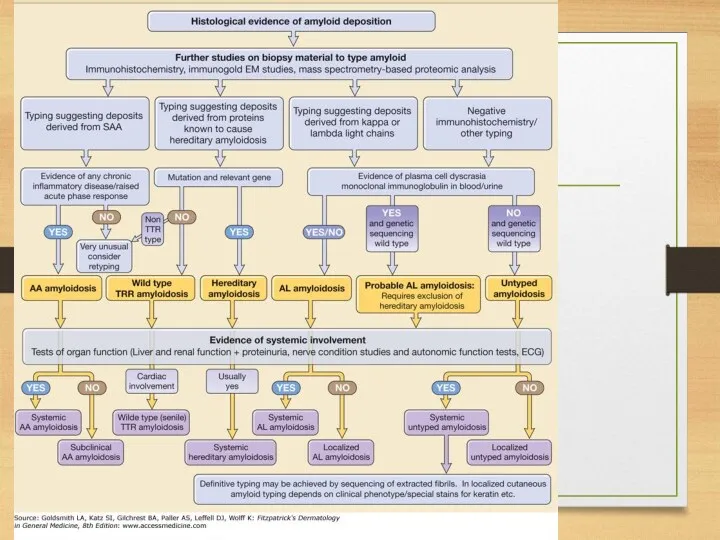
Diagnosis of amyloidosis
1. Presence of amyloid: congo red staining
2. Type
Diagnosis of amyloidosis
1. Presence of amyloid: congo red staining
2. Type

Tissues for biopsy
subcutaneous fat aspiration (provides enough material for all investigations)
Tissues for biopsy
subcutaneous fat aspiration (provides enough material for all investigations)

AA
SAA precursor level in blood
Serum immunoglobulins (to exclude AL;in AA amyloidosis
AA
SAA precursor level in blood
Serum immunoglobulins (to exclude AL;in AA amyloidosis

Instrumental methods
Avoid IV pyelography if amyloidosis is suspected (more frequent renal
Instrumental methods
Avoid IV pyelography if amyloidosis is suspected (more frequent renal
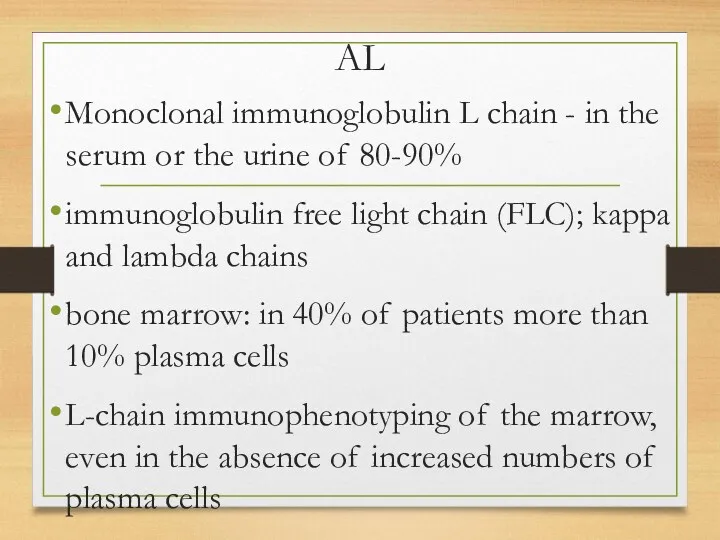
AL
Monoclonal immunoglobulin L chain - in the serum or the urine
AL
Monoclonal immunoglobulin L chain - in the serum or the urine

Biochemistry
concentration of normal Ig is often decreased
hypogammaglobulinemia + proteinuria suggests a
Biochemistry
concentration of normal Ig is often decreased
hypogammaglobulinemia + proteinuria suggests a

Functional systems tests
clotting system abnormalities
kidney function tests
liver function tests
Functional systems tests
clotting system abnormalities
kidney function tests
liver function tests
Instrumental
Echocardiography
Radiolabeled pentagonal (P) component scanning: total body burden of amyloid
Bone
Instrumental
Echocardiography
Radiolabeled pentagonal (P) component scanning: total body burden of amyloid
Bone

ATTR
subcutaneous fat aspiration
sural nerve biopsy
rectum, stomach, myocardium biopsy
Congo red; antiserum
ATTR
subcutaneous fat aspiration
sural nerve biopsy
rectum, stomach, myocardium biopsy
Congo red; antiserum

Instrumental
Echocardiography
Nerve conduction studies to monitor course of disease and assess response
Instrumental
Echocardiography
Nerve conduction studies to monitor course of disease and assess response

Familial systemic (renal)
Biopsy: amyloid confirmation
Affection of organs
SAP component scintigraphy; iodine
Familial systemic (renal)
Biopsy: amyloid confirmation
Affection of organs
SAP component scintigraphy; iodine

beta-2-microglobulin
reference range of serum beta-2-microglobulin concentration of is 1.5-3 mg/L; can
beta-2-microglobulin
reference range of serum beta-2-microglobulin concentration of is 1.5-3 mg/L; can

Radiologic:
joint erosions (usually large joints)
lytic and cystic bone lesions (typically
Radiologic:
joint erosions (usually large joints)
lytic and cystic bone lesions (typically

CT
amyloid deposits: intermediate attenuation.
identification pseudotumors and pseudocystic areas in the
CT
amyloid deposits: intermediate attenuation.
identification pseudotumors and pseudocystic areas in the

MRI
differentiating destructive spondyloarthropathies from inflammatory processes and infections.
MRI
differentiating destructive spondyloarthropathies from inflammatory processes and infections.

Ultrasound
tendon thickness.
rotator cuff thickness greater than 8 mm, thickening of
Ultrasound
tendon thickness.
rotator cuff thickness greater than 8 mm, thickening of

Scintigraphy
radiolabeled P-component scans:
iodine I 123 serum amyloid P
iodohippurate sodium
Scintigraphy
radiolabeled P-component scans:
iodine I 123 serum amyloid P
iodohippurate sodium

Biopsy with Congo red staining and with immunostaining
centrifuged synovial fluid
Biopsy with Congo red staining and with immunostaining
centrifuged synovial fluid
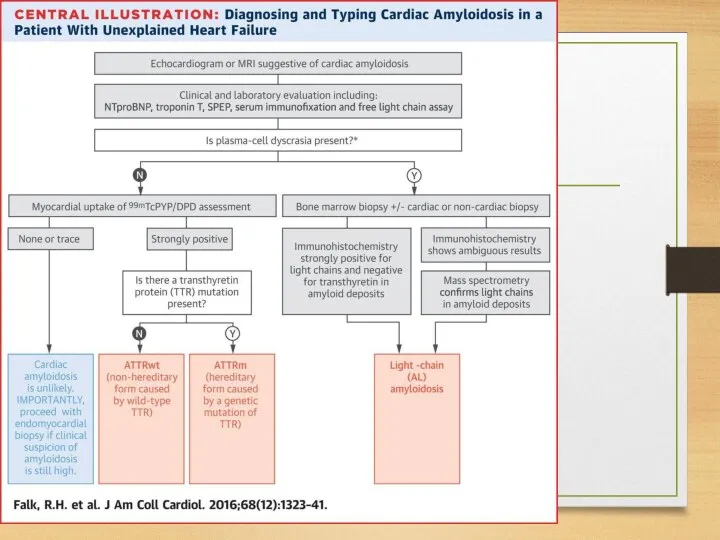
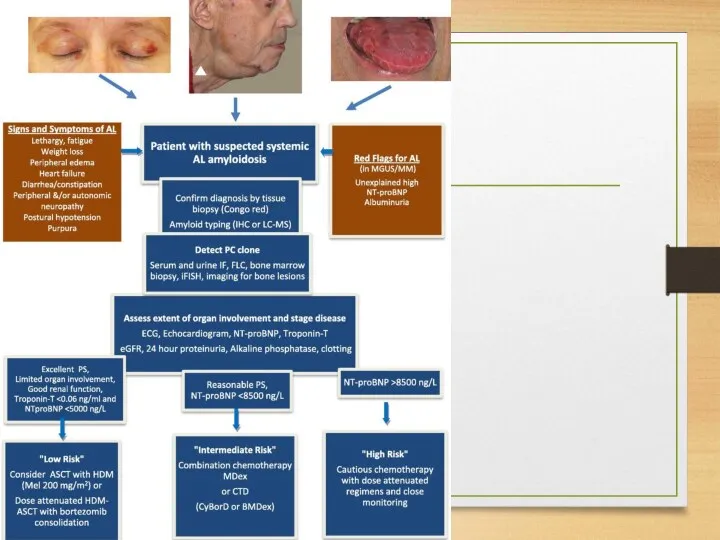

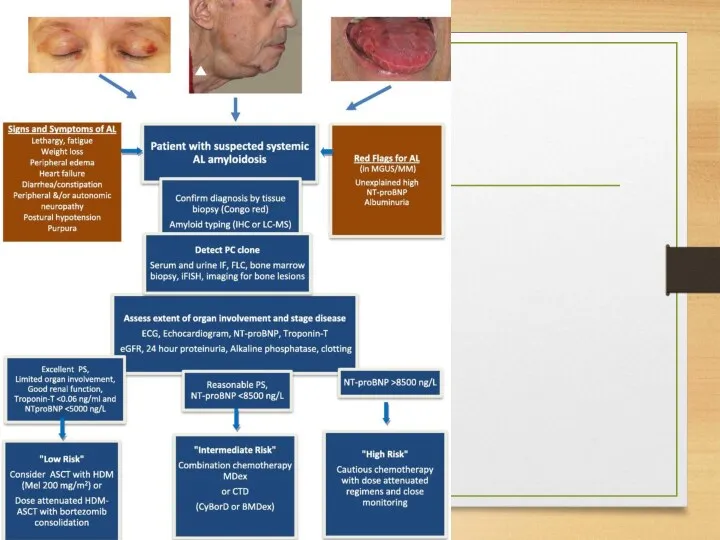
Treatment: AA
primary inflammatory disease treatment
tumor necrosis factor-a inhibitors and interleukin-1 inhibitors
Treatment: AA
primary inflammatory disease treatment
tumor necrosis factor-a inhibitors and interleukin-1 inhibitors
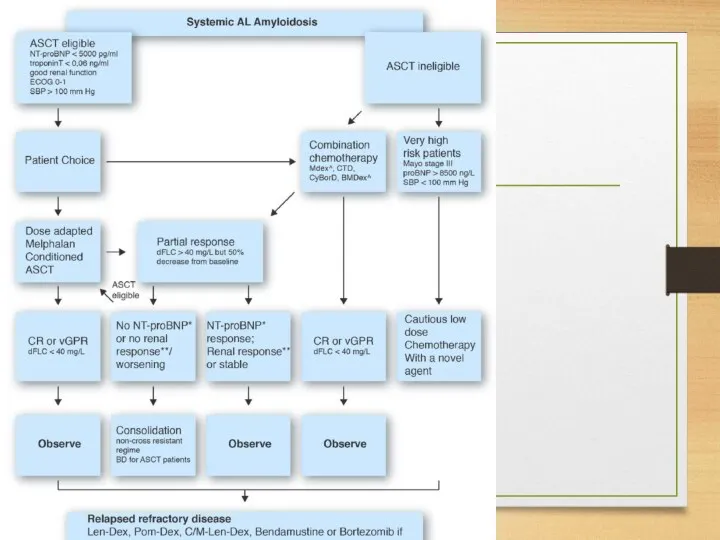

melphalan plus prednisone
melphalan, prednisone, and colchicine
Other chemotherapeutic regimens used
melphalan plus prednisone
melphalan, prednisone, and colchicine
Other chemotherapeutic regimens used

Treatment of localized amyloid L-chain type
has not been studied systematically
chemotherapy
Treatment of localized amyloid L-chain type
has not been studied systematically
chemotherapy

TTR
Digoxin and calcium channel blockers are contraindicated
Liver transplantation
patients with
TTR
Digoxin and calcium channel blockers are contraindicated
Liver transplantation
patients with

beta-2-microglobulin
no adequate treatment (symptomatic)
Improvement of dialysis membranes
Online hemodiafiltration
Direct
beta-2-microglobulin
no adequate treatment (symptomatic)
Improvement of dialysis membranes
Online hemodiafiltration
Direct
 The morphology and reactivity of model catalysts based on cobalt oxide
The morphology and reactivity of model catalysts based on cobalt oxide церкви вологды
церкви вологды Композиции рекламного текста. Первый уровень AIDA
Композиции рекламного текста. Первый уровень AIDA Организация процесса обучения. Содержание
Организация процесса обучения. Содержание Питание и пищеварение. В чем состоит значение питания. Какие системы обеспечивают питание
Питание и пищеварение. В чем состоит значение питания. Какие системы обеспечивают питание Семинар-практикум Обновление предметно-развивающей среды в группах в соответствии с внедрением в образовательный процесс ФГТ
Семинар-практикум Обновление предметно-развивающей среды в группах в соответствии с внедрением в образовательный процесс ФГТ Урок математики в 6 классе Координаты на прямой
Урок математики в 6 классе Координаты на прямой Валерий Яковлевич Брюсов (1873 —1924)
Валерий Яковлевич Брюсов (1873 —1924) Основные клинические классификации нарушений интеллектуального развития
Основные клинические классификации нарушений интеллектуального развития Систематизация и обеспечение сохранности документов. Номенклатура дел организаци
Систематизация и обеспечение сохранности документов. Номенклатура дел организаци Социально-экономическое развитие Руси в XI веке
Социально-экономическое развитие Руси в XI веке Методическое объединение.
Методическое объединение. Среднее арифметическое. Среднее значение величины
Среднее арифметическое. Среднее значение величины Дождевой червь
Дождевой червь Строительно-монтажные работы при замене лифтового оборудования
Строительно-монтажные работы при замене лифтового оборудования терминальные состояния
терминальные состояния Проект Пой, гитара звонкая
Проект Пой, гитара звонкая Николай Николаевич Носов. Федина задача
Николай Николаевич Носов. Федина задача Концепция свадьбы по мотивам фильма В живых останутся только любовники
Концепция свадьбы по мотивам фильма В живых останутся только любовники Основы проектирования и оборудование
Основы проектирования и оборудование Служба безопасности
Служба безопасности Олимпиала в Сочи 2014
Олимпиала в Сочи 2014 Класс Однодольные. Семейство Злаки
Класс Однодольные. Семейство Злаки Светлый праздник Пасхи
Светлый праздник Пасхи Match the verbs to the nouns
Match the verbs to the nouns Тайна имени
Тайна имени Коралові острови
Коралові острови Совместное использование сигналов GPS и ГЛОНАСС
Совместное использование сигналов GPS и ГЛОНАСС