- Генетика рака

Содержание
- 2. Mutations: Somatic and Germline Mutation in egg or sperm Nonheritable Somatic mutations Occur in nongermline tissues
- 3. Tumors Are Clonal Malignant cells
- 4. Somatic Mutations Diabetic islet cell Normal islet cell Normal lung cell Lung cancer cell Many years
- 5. De Novo Mutations New mutation in germ cell No family history of hereditary cancer De novo
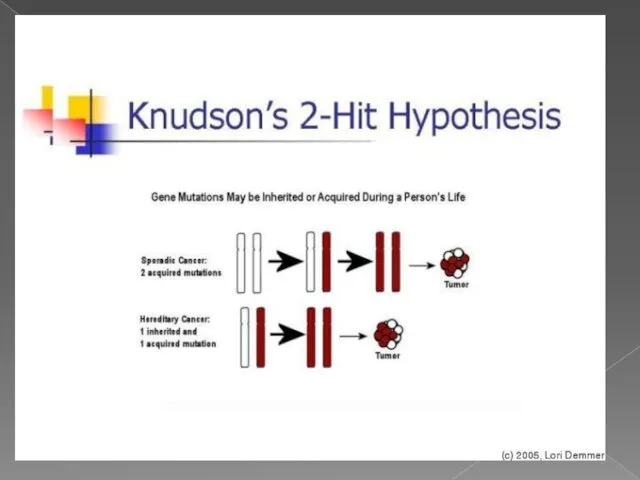
- 6. теория двойного удара или двойной мутации В 1971 году Альфред Кнудсон предложил гипотезу, известную сейчас как
- 9. ОНКОГЕН — это ген, продукт которого может стимулировать образование злокачественной опухоли. Мутации, вызывающие активацию онкогенов, повышают
- 10. Протоонкоген — это обычный ген, который может стать онкогеном из-за мутаций или повышения экспрессии. Многие протоонкогены
- 11. Протоонкоген может стать онкогеном путем относительно незначительной модификации его естественной функции. три основных пути активации: 1.
- 12. Abnormal Cell Growth: Oncogenes Proto-oncogene to oncogene 1st mutation (leads to accelerated cell division) Normal genes
- 13. Tumor Suppressor Genes 1st mutation (leads to accelerated cell division) Normal genes (regulate cell growth) Tumor
- 14. Mutations in Tumor Suppressor Genes 1st mutation (susceptible carrier) Active oncogene No brakes! Active oncogene Normal
- 15. Two-Hit Hypothesis If first hit is a germline mutation, second somatic mutation more likely to enable
- 16. Regulatory Mutations Chromosome 17 Messenger RNA Her2 gene Her2 gene amplification Overexpression Her2 protein Her2 protein
- 17. Translocation of Bcr-Abl Genes Fusion protein with tyrosine kinase activity (q+) Ph (22q–) bcr-abl abl bcr
- 18. Different Locus, Different Allele, Same Phenotype Chromosome 17 BRCA1 BRCA2 Locus (spot on gene) Allele (gene)
- 19. Founder Effect in Ashkenazi Jewish Population An estimated 1 in 40 Ashkenazi Jews carries a BRCA1
- 20. Mutations in Cancer Susceptibility Genes: BRCA1 Nonsense/Frameshift Missense Splice-site Protein has role in genomic stability ~500
- 21. Mutations in Cancer Susceptibility Genes: BRCA2 Nonsense/Frameshift Missense Protein has role in genomic stability ~300 different
- 22. Autosomal Dominant Inheritance Equally transmitted by men and women No skipped generations Each child has a
- 23. Examples of Dominantly Inherited Cancer Syndromes
- 24. Autosomal Recessive Inheritance Two germline mutations (one from each parent) to develop disease Equally transmitted by
- 25. Some Recessively Inherited Cancer Syndromes
- 26. Other Genetic Conditions Linked to Increased Cancer Risk
- 27. Repair Failure Cancer Aging Inborn disease (Transient) cell cycle arrest Apoptosis (cell death) Nucleotide-excision repair (NER)
- 28. Cancer Susceptibility: Much Still Unknown
- 29. How do people know if they should consider genetic testing for BRCA1 and BRCA2 mutations? For
- 30. Li-Fraumeni Syndrome Li-Fraumeni Syndrome (LFS) was first described in 1969 by Drs. Frederick Li and Joseph
- 31. Classic Li-Fraumeni Syndrome (LFS): Three features must be present in a family to fit the classic
- 32. Li-Fraumeni-Like Syndrome (LFL): A person with any childhood cancer or sarcoma, brain tumor, or adrenal cortical
- 33. What Causes LFS? Changes in a “tumor suppressor” gene called “TP53” were discovered in 1990 as
- 34. Risk of Cancer in Patients with LFS The lifetime risk of cancer – all types combined
- 35. For now, in persons with a TP53 gene mutation, we can try to find cancers as
- 36. Cowden syndrome mutations in the PTEN gene Cowden syndrome is a disorder characterized by multiple noncancerous,
- 37. Cowden syndrome mutations in the PTEN gene (TSG) Cowden syndrome is associated with an increased risk
- 38. Что такое ОНКОГЕН ? 1.ген, стимулирующий образование опухоли 2. гены, предохраняющие клетки от ракового перерождения 3.
- 40. Скачать презентацию
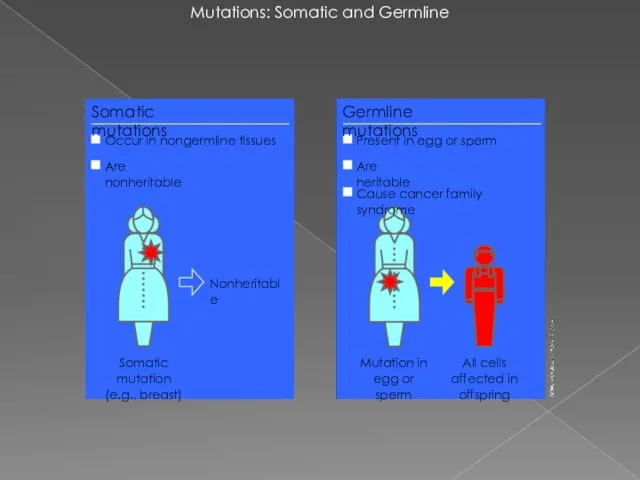
Mutations: Somatic and Germline
Mutation in egg or sperm
Nonheritable
Somatic mutations
Occur in nongermline
Mutations: Somatic and Germline
Mutation in egg or sperm
Nonheritable
Somatic mutations
Occur in nongermline
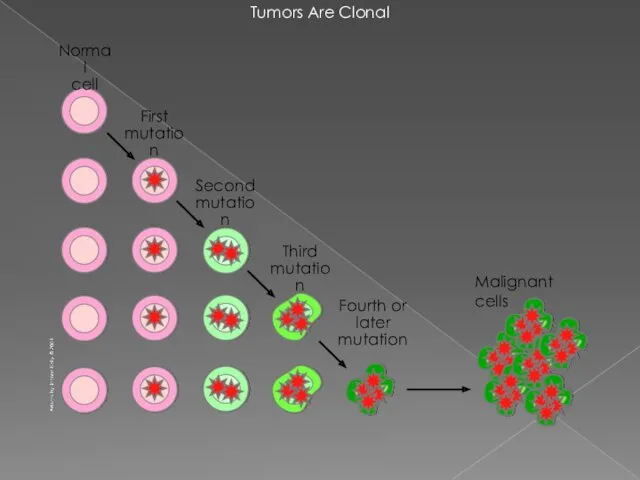
Tumors Are Clonal
Malignant cells
Tumors Are Clonal
Malignant cells
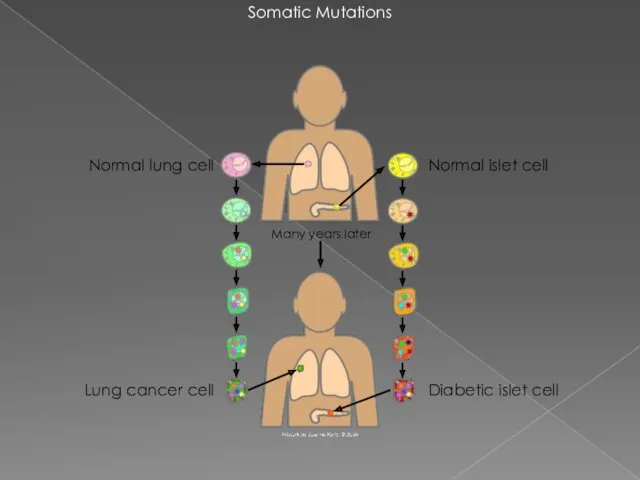
Somatic Mutations
Diabetic islet cell
Normal islet cell
Normal lung cell
Lung cancer cell
Many years
Somatic Mutations
Diabetic islet cell
Normal islet cell
Normal lung cell
Lung cancer cell
Many years
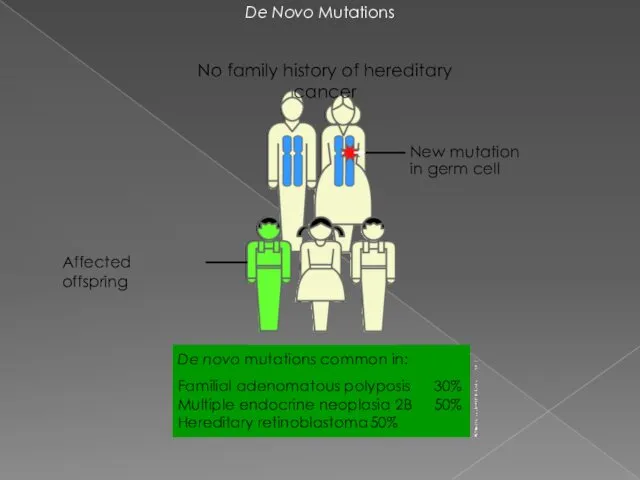
De Novo Mutations
New mutation in germ cell
No family history of hereditary
De Novo Mutations
New mutation in germ cell
No family history of hereditary

теория двойного удара или двойной мутации
В 1971 году Альфред Кнудсон предложил
теория двойного удара или двойной мутации
В 1971 году Альфред Кнудсон предложил


ОНКОГЕН — это ген, продукт которого может стимулировать образование злокачественной опухоли.
ОНКОГЕН — это ген, продукт которого может стимулировать образование злокачественной опухоли.

Протоонкоген — это обычный ген, который может стать онкогеном из-за мутаций
Протоонкоген — это обычный ген, который может стать онкогеном из-за мутаций

Протоонкоген может стать онкогеном путем относительно незначительной модификации его естественной функции.
Протоонкоген может стать онкогеном путем относительно незначительной модификации его естественной функции.
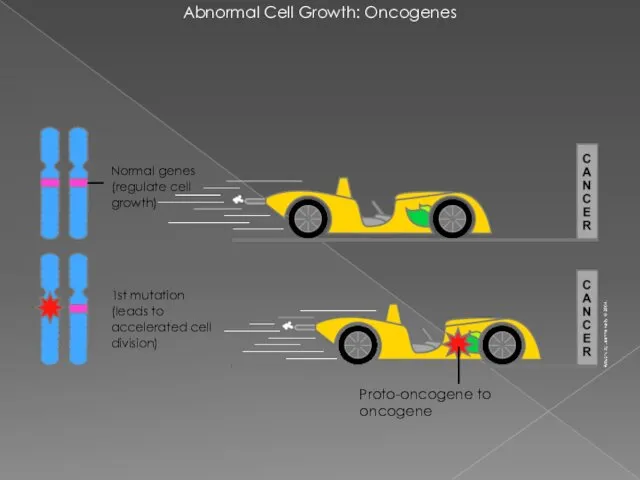
Abnormal Cell Growth: Oncogenes
Proto-oncogene to oncogene
1st mutation
(leads to accelerated cell
Abnormal Cell Growth: Oncogenes
Proto-oncogene to oncogene
1st mutation (leads to accelerated cell
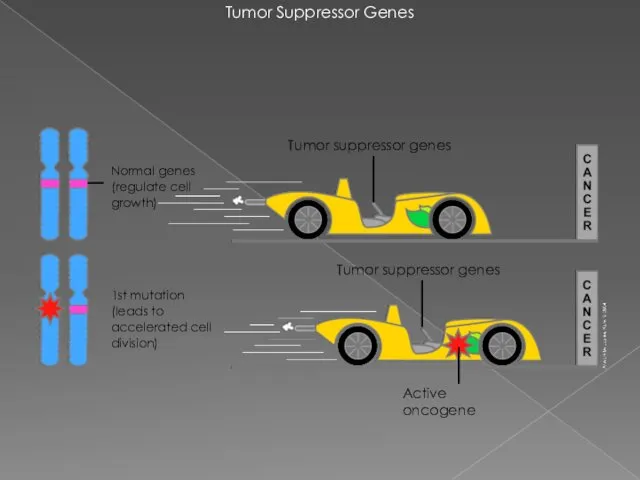
Tumor Suppressor Genes
1st mutation
(leads to accelerated cell division)
Normal genes (regulate
Tumor Suppressor Genes
1st mutation
(leads to accelerated cell division)
Normal genes (regulate

Mutations in Tumor Suppressor Genes
1st mutation (susceptible carrier)
Active oncogene
No brakes!
Active oncogene
Normal
Mutations in Tumor Suppressor Genes
1st mutation (susceptible carrier)
Active oncogene
No brakes!
Active oncogene
Normal
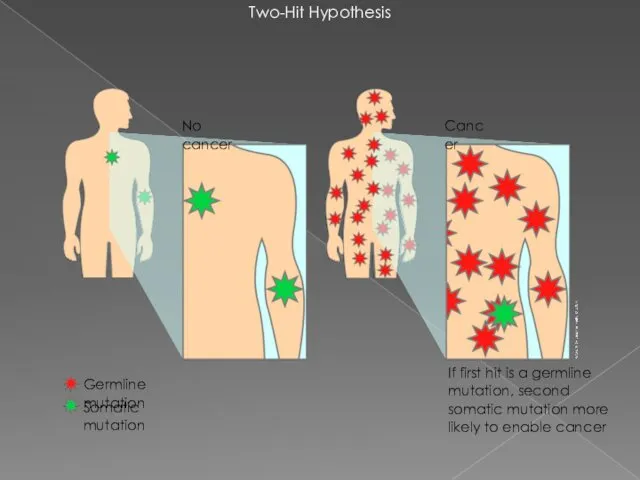
Two-Hit Hypothesis
If first hit is a germline mutation, second somatic mutation
Two-Hit Hypothesis
If first hit is a germline mutation, second somatic mutation
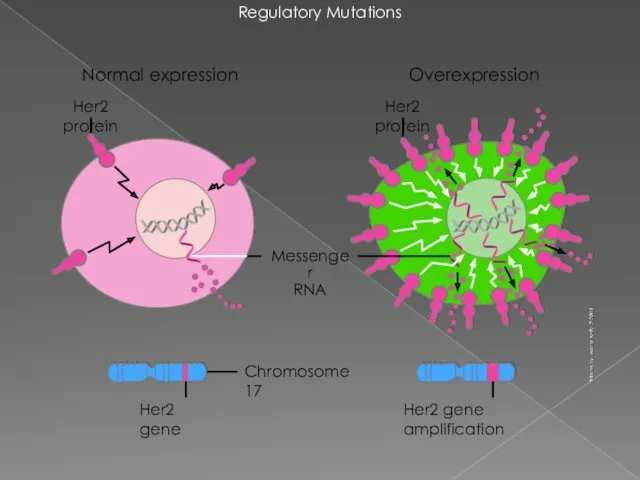
Regulatory Mutations
Chromosome 17
Messenger
RNA
Her2 gene
Her2 gene amplification
Overexpression
Her2 protein
Her2 protein
Normal expression
Regulatory Mutations
Chromosome 17
Messenger
RNA
Her2 gene
Her2 gene amplification
Overexpression
Her2 protein
Her2 protein
Normal expression

Translocation of Bcr-Abl Genes
Fusion protein with tyrosine kinase activity
(q+)
Ph
(22q–)
bcr-abl
abl
bcr
22
9
9
Translocation of Bcr-Abl Genes
Fusion protein with tyrosine kinase activity
(q+)
Ph
(22q–)
bcr-abl
abl
bcr
22
9
9
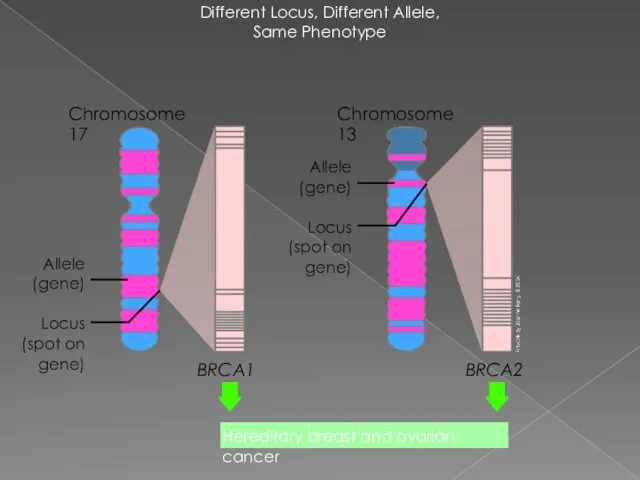
Different Locus, Different Allele,
Same Phenotype
Chromosome 17
BRCA1
BRCA2
Locus (spot on gene)
Allele (gene)
Chromosome 13
Hereditary
Different Locus, Different Allele,
Same Phenotype
Chromosome 17
BRCA1
BRCA2
Locus (spot on gene)
Allele (gene)
Chromosome 13
Hereditary

Founder Effect in
Ashkenazi Jewish Population
An estimated 1 in 40 Ashkenazi Jews
Founder Effect in
Ashkenazi Jewish Population
An estimated 1 in 40 Ashkenazi Jews

Mutations in
Cancer Susceptibility Genes: BRCA1
Nonsense/Frameshift
Missense
Splice-site
Protein has role in genomic stability
~500
Mutations in
Cancer Susceptibility Genes: BRCA1
Nonsense/Frameshift
Missense
Splice-site
Protein has role in genomic stability
~500
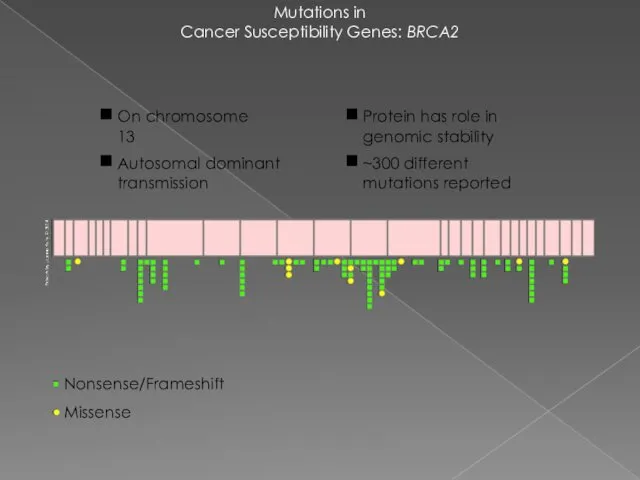
Mutations in
Cancer Susceptibility Genes: BRCA2
Nonsense/Frameshift
Missense
Protein has role in genomic stability
~300 different
Mutations in
Cancer Susceptibility Genes: BRCA2
Nonsense/Frameshift
Missense
Protein has role in genomic stability
~300 different
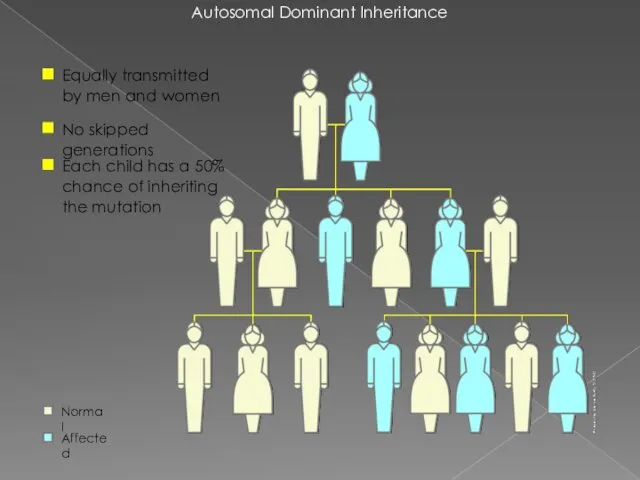
Autosomal Dominant Inheritance
Equally transmitted by men and women
No skipped generations
Each child
Autosomal Dominant Inheritance
Equally transmitted by men and women
No skipped generations
Each child
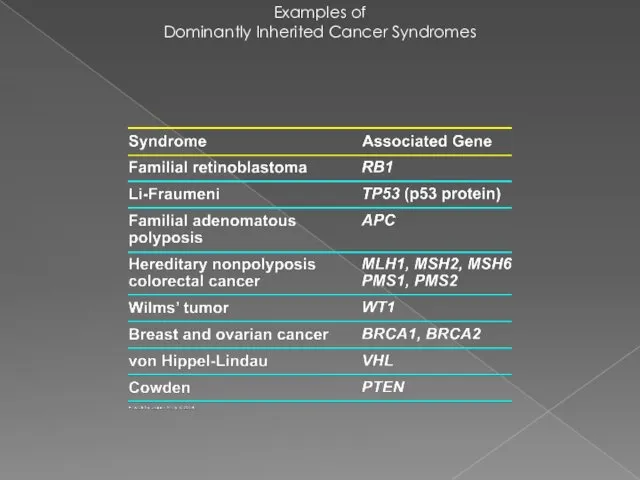
Examples of
Dominantly Inherited Cancer Syndromes
Examples of
Dominantly Inherited Cancer Syndromes
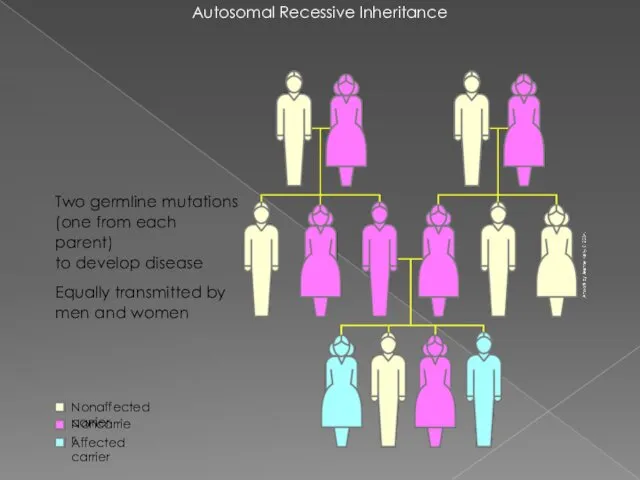
Autosomal Recessive Inheritance
Two germline mutations
(one from each parent)
to develop disease
Equally transmitted
Autosomal Recessive Inheritance
Two germline mutations
(one from each parent)
to develop disease
Equally transmitted

Some Recessively Inherited
Cancer Syndromes
Some Recessively Inherited
Cancer Syndromes

Other Genetic Conditions
Linked to Increased Cancer Risk
Other Genetic Conditions
Linked to Increased Cancer Risk

Repair Failure
Cancer
Aging
Inborn
disease
(Transient) cell cycle arrest
Apoptosis
(cell death)
Nucleotide-excision repair
(NER)
Base-
excision repair (BER)
Mismatch Repair
Recombinational
repair
(HR, EJ)
Uracil
Abasic
site
B-oxoguanine
Single-strand
Repair Failure
Cancer
Aging
Inborn
disease
(Transient) cell cycle arrest
Apoptosis
(cell death)
Nucleotide-excision repair
(NER)
Base-
excision repair (BER)
Mismatch Repair
Recombinational
repair
(HR, EJ)
Uracil
Abasic
site
B-oxoguanine
Single-strand

Cancer Susceptibility:
Much Still Unknown
Cancer Susceptibility:
Much Still Unknown

How do people know if they should consider genetic testing for
How do people know if they should consider genetic testing for

Li-Fraumeni Syndrome
Li-Fraumeni Syndrome (LFS) was first described in 1969 by Drs.
Li-Fraumeni Syndrome
Li-Fraumeni Syndrome (LFS) was first described in 1969 by Drs.

Classic Li-Fraumeni Syndrome (LFS):
Three features must be present in
Classic Li-Fraumeni Syndrome (LFS):
Three features must be present in

Li-Fraumeni-Like Syndrome (LFL):
A person with any childhood cancer or sarcoma,
Li-Fraumeni-Like Syndrome (LFL):
A person with any childhood cancer or sarcoma,

What Causes LFS?
Changes in a “tumor suppressor” gene called “TP53” were
What Causes LFS?
Changes in a “tumor suppressor” gene called “TP53” were

Risk of Cancer in Patients with LFS
The lifetime risk of cancer
Risk of Cancer in Patients with LFS
The lifetime risk of cancer

For now, in persons with a TP53 gene mutation, we can
For now, in persons with a TP53 gene mutation, we can

Cowden syndrome
mutations in the PTEN gene
Cowden syndrome is a disorder
Cowden syndrome
mutations in the PTEN gene
Cowden syndrome is a disorder

Cowden syndrome
mutations in the PTEN gene (TSG)
Cowden syndrome is associated
Cowden syndrome
mutations in the PTEN gene (TSG)
Cowden syndrome is associated

Что такое ОНКОГЕН ?
1.ген, стимулирующий образование опухоли
2. гены, предохраняющие клетки от
Что такое ОНКОГЕН ?
1.ген, стимулирующий образование опухоли
2. гены, предохраняющие клетки от
 Формирование учебных универсальных действий (УУД) во внеурочной деятельности учащихся
Формирование учебных универсальных действий (УУД) во внеурочной деятельности учащихся Индивидуальный проект Государственное устройство Древней Спарты
Индивидуальный проект Государственное устройство Древней Спарты Мои проекты
Мои проекты Эмиссия. Выпуск денег в хозяйственный оборот
Эмиссия. Выпуск денег в хозяйственный оборот Роль логистики в организации
Роль логистики в организации Витебская область
Витебская область Безопасность в сети интернет
Безопасность в сети интернет Классификация барж по типу передвижения
Классификация барж по типу передвижения Картотека зимние подвижные игры
Картотека зимние подвижные игры Конструкция скважины
Конструкция скважины как определить падеж сущ-х
как определить падеж сущ-х Шаблонирование насосно - компрессорных труб (НКТ)
Шаблонирование насосно - компрессорных труб (НКТ) Лексика и фразеология
Лексика и фразеология Общие начала назначения наказания. УКРФ
Общие начала назначения наказания. УКРФ Прогнозирование течения эпилепсии на основе DFA анализа ЭЭГ
Прогнозирование течения эпилепсии на основе DFA анализа ЭЭГ Ты воспитатель, а это значит...
Ты воспитатель, а это значит... Кариес зубов у детей. Лечение
Кариес зубов у детей. Лечение Кейнсианская модель макроэкономического равновесия. (Тема 3)
Кейнсианская модель макроэкономического равновесия. (Тема 3) Франция. Экономическое развитие Франции. Политическое развитие Франции
Франция. Экономическое развитие Франции. Политическое развитие Франции Машины для разделения неоднородных систем
Машины для разделения неоднородных систем Медиакит радио GOLDSTAR
Медиакит радио GOLDSTAR Мировое хозяйство
Мировое хозяйство Компрессор ПК - 5,25 Тепловоза ТГМ-6
Компрессор ПК - 5,25 Тепловоза ТГМ-6 Презентация Наша Масленица
Презентация Наша Масленица Становление народного образования
Становление народного образования Отчёт о прохождении производственной практики
Отчёт о прохождении производственной практики Основные инициативы в области КСО и устойчивого развития. Отчетность компании в области КСО. Аудит отчетности
Основные инициативы в области КСО и устойчивого развития. Отчетность компании в области КСО. Аудит отчетности Всероссийский физкультурно-спортивный комплекс Готов к труду и обороне
Всероссийский физкультурно-спортивный комплекс Готов к труду и обороне