- методы медгенетики2020

Содержание
- 6. Генеалогический метод Генеология – наука о родословных. Технически клинико-генеологический метод складывается из 2 этапов: Составление родословной
- 7. Символика
- 9. Аутосомно-доминантный тип наследования: Экспрессивность – это степень выраженности действия гена у отдельной особи. Понятие экспрессивности аналогично
- 21. Тип наследования, сцепленный с полом: Гемофилия – Виктория. Королева Англии, родив сына, страдающего гемофилией, через своих
- 26. Близнецовый метод Коркондантность – процент сходства по изучаемому признаку. Дискордантность – отсутствие признака у одного из
- 27. Биохимические методы Методы, позволяющие обнаружить целый ряд наследственных заболеваний, причиной которых являются нарушения обмена веществ (энзимопатии),
- 28. Популяционно-статистический метод Популяционная генетика изучает взаимодействие факторов, влияющих на распределение наследственных признаков в популяции. Популяция –
- 29. Цитологический метод Основан на микроскопическом исследовании хромосом, определение специфичности кариотипа. Исследование полового хроматина Половой хроматин –
- 31. Методы генетики соматических клеток Культивирование отдельных соматических клеток и получение клонов, а также их гибридизацию и
- 32. Молекулярно-генетические методы В середине 80-х годов были разработаны методы ДНК-зондовой диагностики, которые позволяют распознать заболевание по
- 33. Методы выявления гетерозиготного носительства у женщин Если отец поражен наследственной болезнью Если женщина родила 2-х и
- 34. Выявление состояния гетерозиготного носительства Клиническое изучение микросимптомов заболевания с выявлением аномалий развития Использование нагрузочных тестов Микроскопическое
- 35. Методы пренатальной диагностики Инвазивные методы Амниоцентез – исследование клеток, белков, гормонов. Химического состава амниотической жидкости Биопсия
- 36. Хромосомными болезнями называются комплексы множественных врожденных пороков развития, вызываемых числовыми (геномные мутации) или структурными (хромосомные аберрации)
- 38. Аномалии аутосом Наиболее часто у человека встречаются трисомии по 21-й, 13-й и 18-й паре хромосом. Синдром
- 39. Факторы риска Возраст матери 35-46 лет (вероятность рождения больного ребенка возрастает до 4,1%). Возможность возникновения повторного
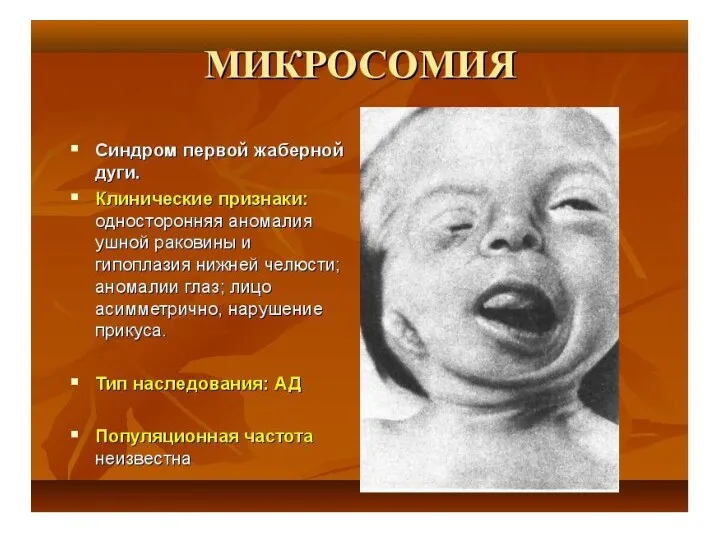
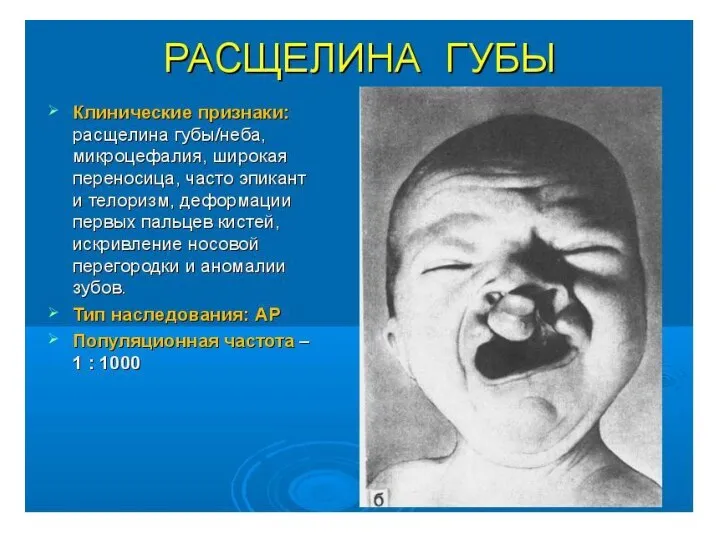
- 40. Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо. Типичны эпикант,
- 41. Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо. Клинические проявления
- 43. Синдром Патау Синдром трисомии 13 - встречается с частотой 1:6000. Между частотой возникновения синдрома Патау и
- 44. Синдром Патау
- 45. Синдром Эдвардса синдром трисомии 18 - встречается с частотой примерно 1:7000. Дети с трисомией 18 чаще
- 49. АНОМАЛИИ ПОЛОВЫХ ХРОМОСОМ Пол будущего ребенка определяется в момент оплодотворения в зависимости от сочетания половых хромосом
- 50. XX- нормальный женский организм. XXX- синдром трисомии X. Частота встречаемости 1:1000. Кариотип 47,ХХХ. В настоящее время
- 51. Синдрому полисемии X присущ значительный полиморфизм. Женский организм с мужеподобным телосложением. Могут быть недоразвиты первичные и
- 52. ХО - синдром Шерешевского-Тернера (моносомия X) Частота встречаемости 1:2000-1:3000. Кариотип45,Х. У 55% девочек с этим синдромом
- 54. XY- нормальный мужской организм. XXY и XXXY- синдром Клайнфелтера Частота встречаемости 1:500. Кариотип 47,XXY у 80%
- 55. Фенотип мужской. Клиника отличается широким разнообразием и неспецифичностью проявлений. У мальчиков рост превышает средние показатели, характерные
- 57. XXY и XXXY- синдром Клайнфелтера Иногда эффективно раннее лечение мужскими половыми гормонами. Чем больше в наборе
- 58. Болезнь Помпе :
- 59. Болезнь Помпе Прогрессирующее полисистемное , инвалидизирующее, нередко, фатальное нервно-мышечное заболевание Болезнь накопления гликогена (гликогеноз) II типа
- 60. Патогенез болезни Помпе Лизосомный фермент GAA (кислой альфа-глюкозидазы) необходим для разрушения лизосомного гликогена Врожденный дефицит фермента
- 61. Генетика и частота Болезнь Помпе моногенное заболевание Ген GAA локализован в длинном плече 17 хромосомы (17q25)
- 62. Болезнь Помпе Характеризуется дефицитом лизосомального фермента кислой альфа-мальтазы (глюкозидазы) (GAA) Как следствие, происходит прогрессирующее внутриклеточное накопление
- 63. Различная скорость прогрессирования болезни Течение болезни Помпе Быстро прогрессирующее Медленно прогрессирующее
- 64. Скелетные мышцы Глубокая и быстро прогрессирующая мышечная слабость Мышечная гипотония Амиотония Запрокидывание головы Значительно повышенная сывороточная
- 65. Инфантильная форма
- 66. Ювенильная форма
- 67. БОЛЕЗНЬ ПОМПЕ У ДЕТЕЙ И ВЗРОСЛЫХ Дыхательная система Дыхательная недостаточность/дистресс синдром Слабость диафрагмы Нарушения дыхания во
- 68. Сердце Рентгенограмма грудной клетки Электрокардиография (ЭКГ) Эхокардиография (Эхо-КГ) Легкие Спирометрия (оценка ЖЕЛ сидя, лежа) Рентгенограмма грудной
- 69. Мышцы Электромиография (ЭМГ)/исследования нервной проводимости Тестирование мышечной силы Лабораторные исследования Сывороточная креатинкиназа (КК) Аланиновая и аспарагиновая
- 70. ДИАГНОСТИКА Инвазивные методы: биопсия кожи биопсия мышцы В настоящее время анализ сухих пятен крови позволяет с
- 71. ЭЛЕКТРОННАЯ МИКРОСКОПИЯ Здоровые миофибриллы замещаются гликогеном, что постепенно нарушает мышечную функцию Изменения структуры мышц могут предшествовать
- 72. ЛЕЧЕНИЕ Ферментнозамещающая терапия – рекомбинантной человеческой альфа – глюкозидазой (МИОЗИМ) - 20 / 40 мг/кг еженедельно
- 73. Результаты лечения
- 74. РЕЗУЛЬТАТЫ ЛЕЧЕНИЯ Уменьшение потребности в ИВЛ Увеличение ЖЕЛ, возрастание мобильности и способности передвигаться Обретение утраченных двигательных
- 75. БОЛЕЗНЬ ФАБРИ
- 76. Болезнь Фабри Наследственный дефект фермента α-галактозидазы А, приводящий к прогрессерующему накоплению гликосфинголипидов, в основном, в лизосомах
- 77. Фенотип пациентов с БФ
- 78. ОСНОВНЫЕ ПРИЗНАКИ Увеличены нос, уши, язык, слюнные железы Диспропорциональный рост костей черепа (увеличение скуловых костей, надбровных
- 79. Тип наследования– Х-сцепленный рецессивный
- 80. По данным регистра пациентов с БФ между временем появления первых симптомов и датой установления диагноза БФ
- 81. ОРГАНЫ - МИШЕНИ
- 82. Патология кожных покровов: Ангиокератома (71%) Гипогидроз
- 83. Ангиокератома
- 84. Патология периферической нервной системы кризы Фабри –жгучие боли в ладонях и стопах (77%) акропарестезии
- 85. Патология глаз: Помутнение роговицы Катаракта Фабри
- 86. Патология глаз: Извитость и аневризмы сосудов сетчатки Ретинопатия слепота
- 87. Патология сердца: Гипертрофия левого желудочка (88%) Недостаточность митрального клапана Нарушения ритма и проводимости Стенокардия Инфаркт миокарда
- 88. Цереброваскулярная недостаточность Транзиторные ишемические атаки (гемиплегия, гемианестезия, афазия, судороги) Инсульты с характерными изменениями КТ/МРТ На поздних
- 89. Ишемический инсульт
- 90. Патология почек: Протеинурия (84%) Тубулярная дисфункция Высокий уровень креатинина Почечная недостаточность (47%)
- 91. Неспецифические проявления БФ Нейросенсорная тугоухость Хронические обструктивные заболевания бронхов Патология ЖКТ: тошнота, рвота, абдоминальные боли Варикозное
- 92. Лабораторные данные Анализ мочи:протеинурия, гематурия, цилиндрурия, эпителий почечных канальцев, липидные глобулы в виде “мальтийского креста” Анализ
- 93. Традиционная диагностика ЛБН Измерение активности ферментов в: Лейкоцитах, изолированных из крови с ЭДТА Лимфоцитах, изолированных из
- 94. Преимущества метода DBS Позволяет осуществлять пересылку образцов крови Небольшой объем крови для исследования Высушенные пятна крови
- 95. Патогенетическая терапия при болезни Фабри Трансплантация стволовых кроветворных клеток Трансплантация печени плода Геннотерапия Энзимо-заместительная терапия (фабризим)
- 96. Фабразим (агалзидаза бета) – человеческая рекомбинантная альфа-галактозидаза А полностью идентичная нативному ферменту. Специфическая активность – 70
- 97. Фабразим (агалзидаза бета) Флаконы – 35мг и 5 мг Назначается в дозе 1 мг/кг веса Вводится
- 99. Скачать презентацию


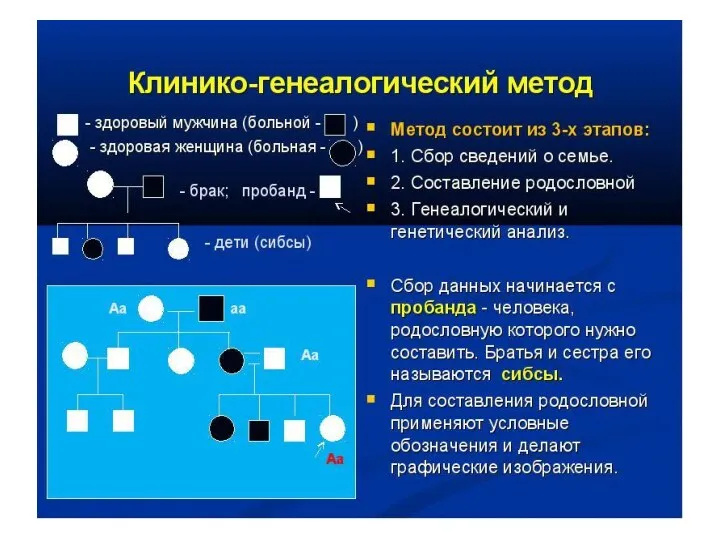


Генеалогический метод
Генеология – наука о родословных.
Технически клинико-генеологический метод складывается из 2
Генеалогический метод
Генеология – наука о родословных.
Технически клинико-генеологический метод складывается из 2
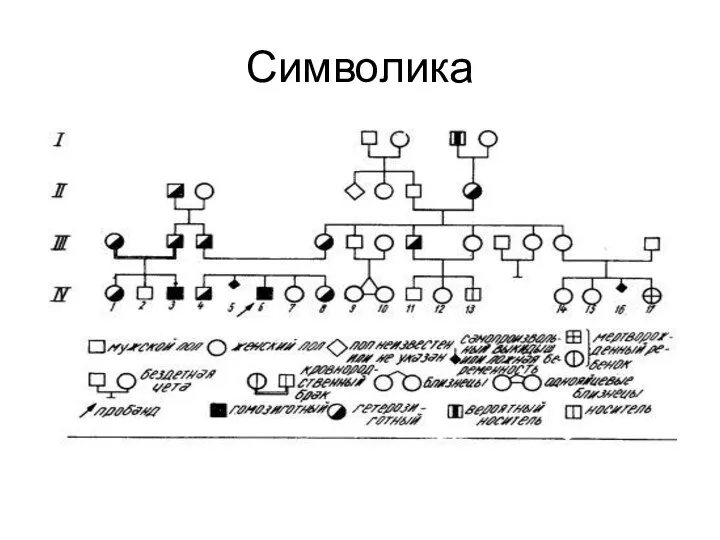
Символика
Символика
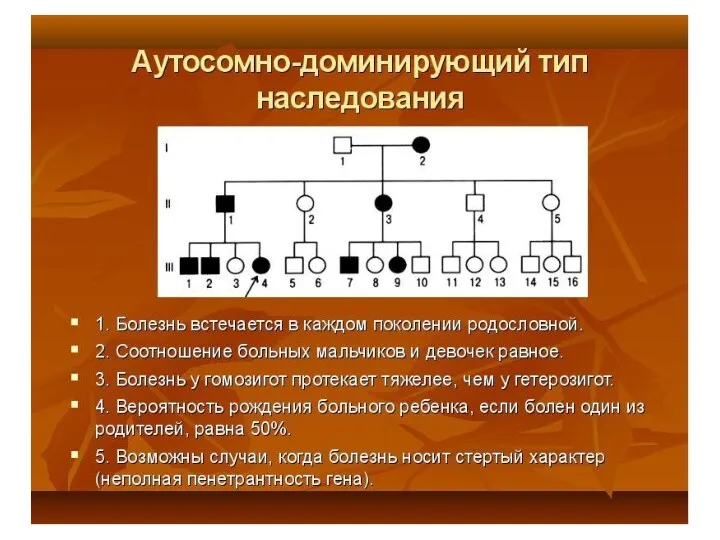

Аутосомно-доминантный тип наследования:
Экспрессивность – это степень выраженности действия гена у отдельной
Аутосомно-доминантный тип наследования:
Экспрессивность – это степень выраженности действия гена у отдельной



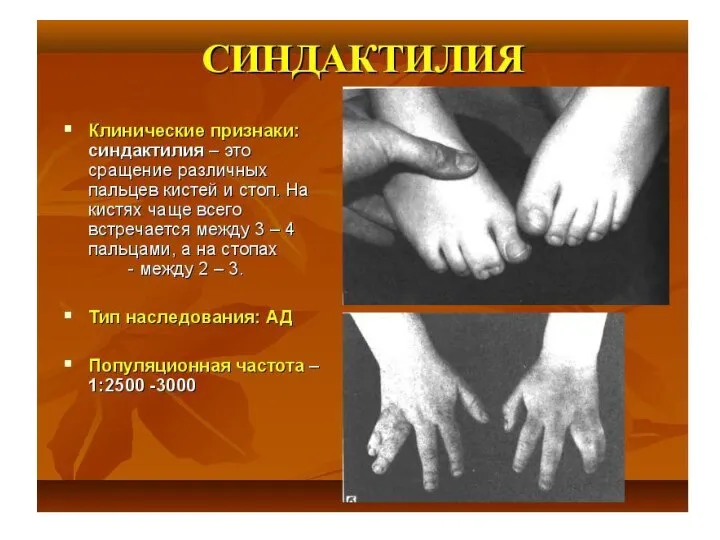
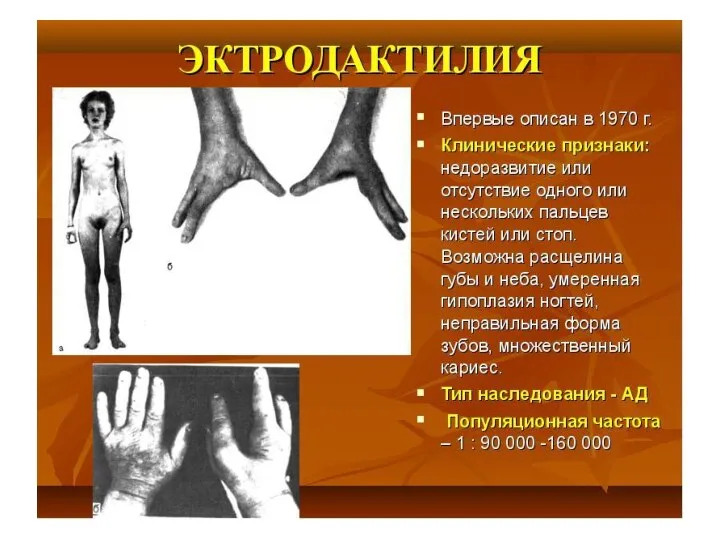
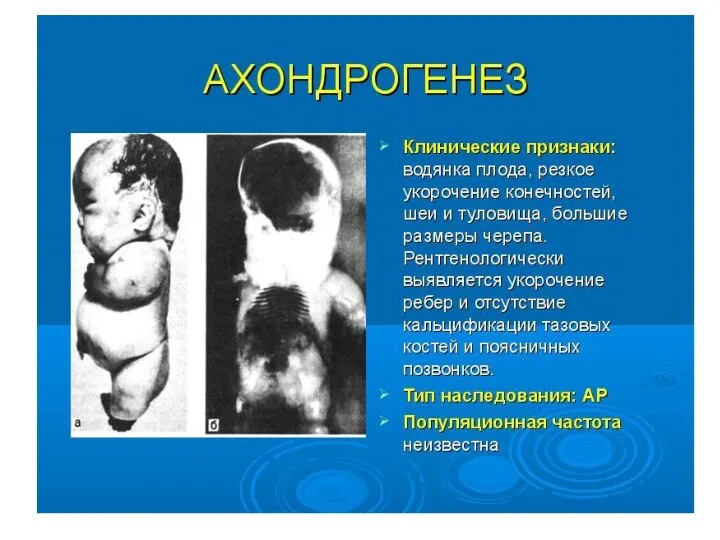

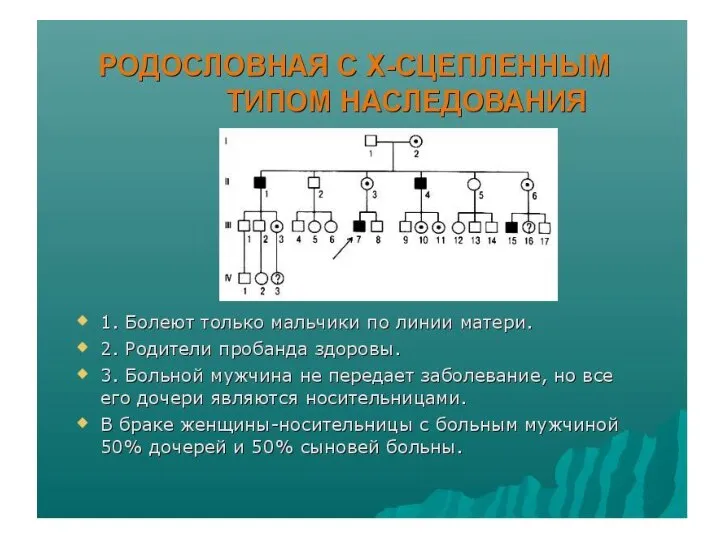

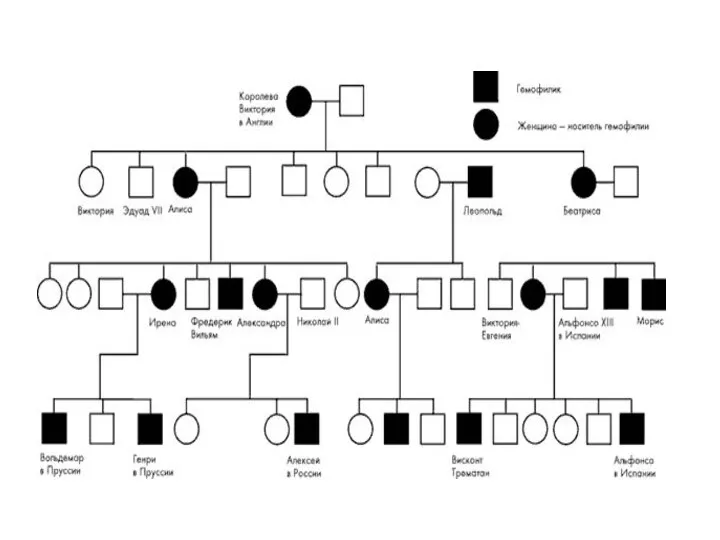
Тип наследования, сцепленный с полом:
Гемофилия – Виктория.
Королева Англии, родив сына,
Тип наследования, сцепленный с полом:
Гемофилия – Виктория.
Королева Англии, родив сына,


Близнецовый метод
Коркондантность – процент сходства по изучаемому признаку.
Дискордантность – отсутствие признака
Близнецовый метод
Коркондантность – процент сходства по изучаемому признаку.
Дискордантность – отсутствие признака

Биохимические методы
Методы, позволяющие обнаружить целый ряд наследственных заболеваний, причиной которых являются
Биохимические методы
Методы, позволяющие обнаружить целый ряд наследственных заболеваний, причиной которых являются

Популяционно-статистический метод
Популяционная генетика изучает взаимодействие факторов, влияющих на распределение наследственных признаков
Популяционно-статистический метод
Популяционная генетика изучает взаимодействие факторов, влияющих на распределение наследственных признаков

Цитологический метод
Основан на микроскопическом исследовании хромосом, определение специфичности кариотипа.
Исследование полового хроматина
Половой
Цитологический метод
Основан на микроскопическом исследовании хромосом, определение специфичности кариотипа.
Исследование полового хроматина
Половой


Методы генетики соматических клеток
Культивирование отдельных соматических клеток и получение клонов, а
Методы генетики соматических клеток
Культивирование отдельных соматических клеток и получение клонов, а

Молекулярно-генетические методы
В середине 80-х годов были разработаны методы ДНК-зондовой диагностики, которые
Молекулярно-генетические методы
В середине 80-х годов были разработаны методы ДНК-зондовой диагностики, которые

Методы выявления гетерозиготного носительства у женщин
Если отец поражен наследственной болезнью
Если женщина
Методы выявления гетерозиготного носительства у женщин
Если отец поражен наследственной болезнью
Если женщина

Выявление состояния гетерозиготного носительства
Клиническое изучение микросимптомов заболевания с выявлением аномалий развития
Использование
Выявление состояния гетерозиготного носительства
Клиническое изучение микросимптомов заболевания с выявлением аномалий развития
Использование

Методы пренатальной диагностики
Инвазивные методы
Амниоцентез – исследование клеток, белков, гормонов. Химического состава
Методы пренатальной диагностики
Инвазивные методы
Амниоцентез – исследование клеток, белков, гормонов. Химического состава

Хромосомными болезнями
называются комплексы множественных врожденных пороков развития, вызываемых числовыми
(геномные
Хромосомными болезнями
называются комплексы множественных врожденных пороков развития, вызываемых числовыми
(геномные


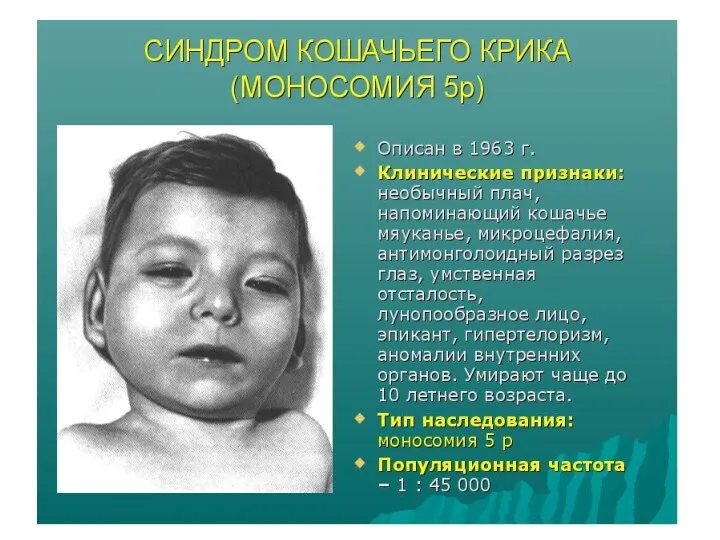
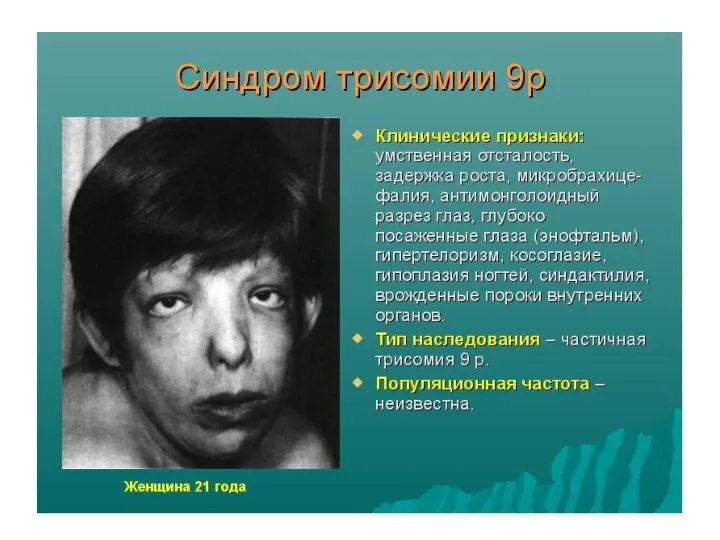
Аномалии аутосом
Наиболее часто у человека встречаются трисомии по 21-й, 13-й и
Аномалии аутосом
Наиболее часто у человека встречаются трисомии по 21-й, 13-й и

Факторы риска
Возраст матери 35-46 лет (вероятность рождения больного ребенка возрастает до
Факторы риска
Возраст матери 35-46 лет (вероятность рождения больного ребенка возрастает до

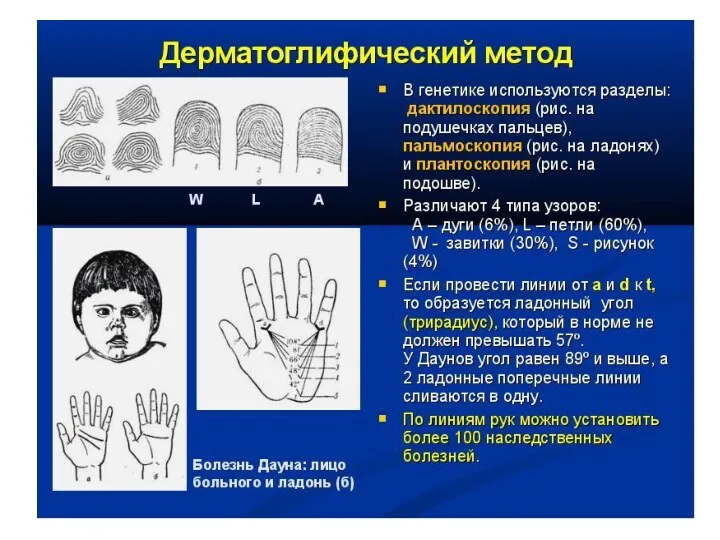
Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб,
Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб,
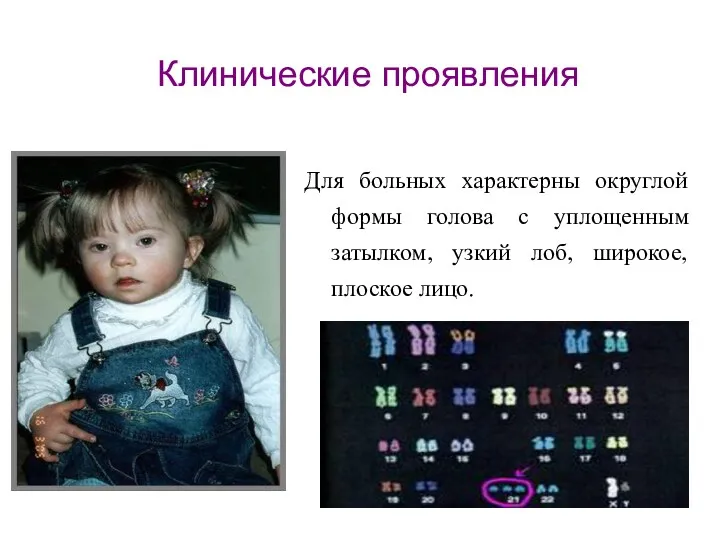
Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб,
Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб,
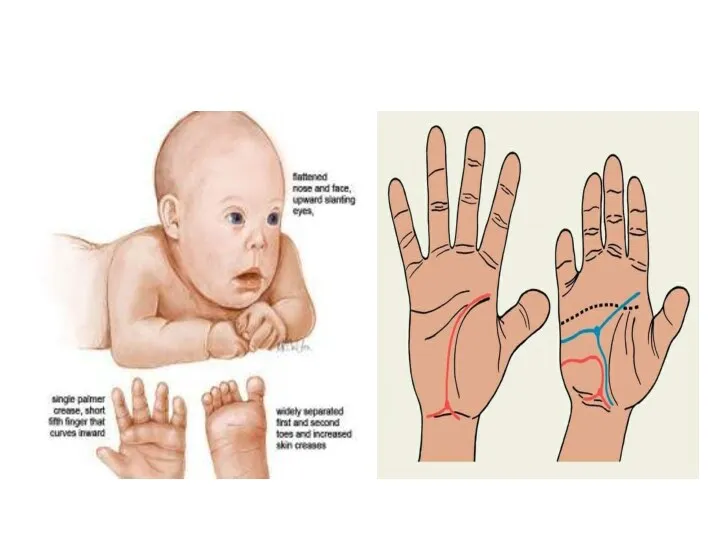

Синдром Патау
Синдром трисомии 13 - встречается с частотой 1:6000.
Синдром Патау
Синдром трисомии 13 - встречается с частотой 1:6000.
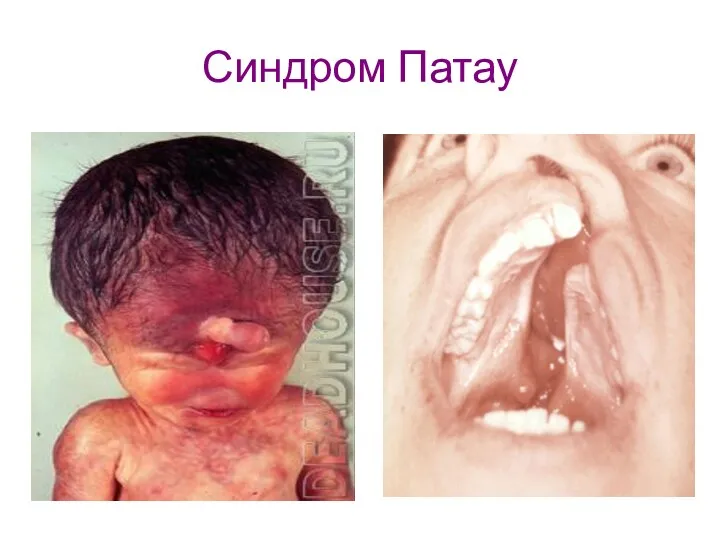
Синдром Патау
Синдром Патау

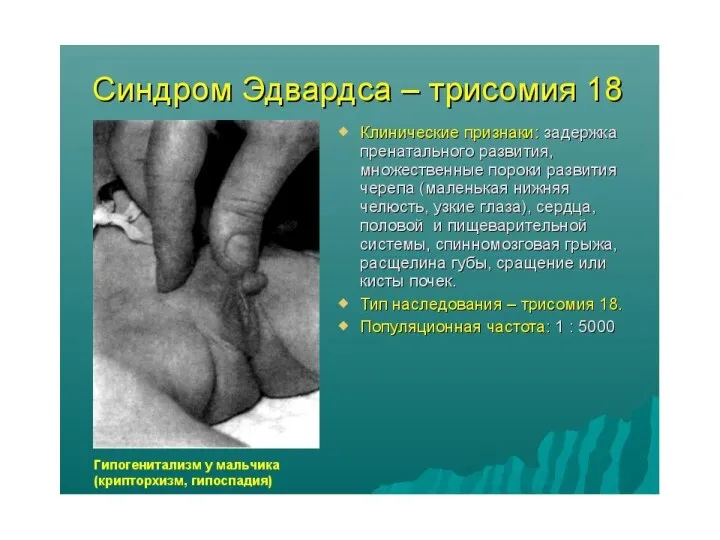
Синдром Эдвардса
синдром трисомии 18 - встречается с частотой примерно 1:7000.
Синдром Эдвардса
синдром трисомии 18 - встречается с частотой примерно 1:7000.

АНОМАЛИИ ПОЛОВЫХ ХРОМОСОМ
Пол будущего ребенка определяется в момент оплодотворения в зависимости
АНОМАЛИИ ПОЛОВЫХ ХРОМОСОМ
Пол будущего ребенка определяется в момент оплодотворения в зависимости

XX- нормальный женский организм.
XXX- синдром трисомии X. Частота встречаемости
XXX- синдром трисомии X. Частота встречаемости

Синдрому полисемии X присущ значительный полиморфизм. Женский организм с мужеподобным телосложением.
Синдрому полисемии X присущ значительный полиморфизм. Женский организм с мужеподобным телосложением.

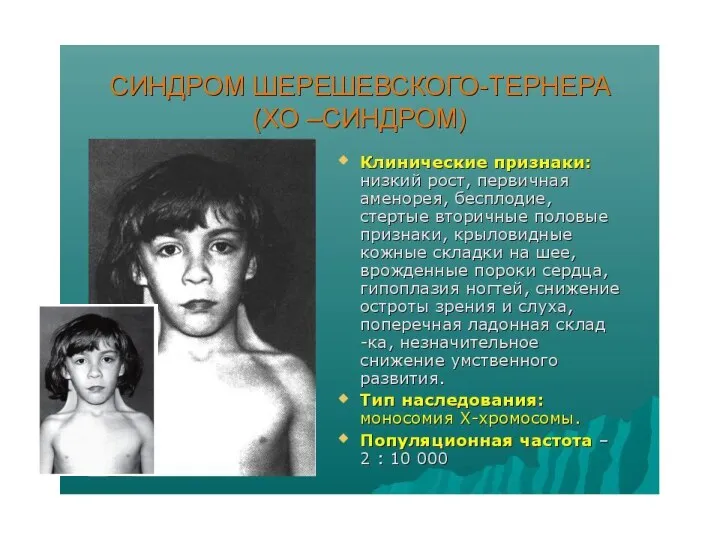
ХО - синдром Шерешевского-Тернера (моносомия X)
Частота встречаемости 1:2000-1:3000. Кариотип45,Х. У
ХО - синдром Шерешевского-Тернера (моносомия X)
Частота встречаемости 1:2000-1:3000. Кариотип45,Х. У

XY- нормальный мужской организм.
XXY и XXXY- синдром Клайнфелтера
Частота встречаемости 1:500.
XY- нормальный мужской организм.
XXY и XXXY- синдром Клайнфелтера
Частота встречаемости 1:500.

Фенотип мужской. Клиника отличается широким разнообразием и неспецифичностью проявлений. У мальчиков
Фенотип мужской. Клиника отличается широким разнообразием и неспецифичностью проявлений. У мальчиков


XXY и XXXY- синдром Клайнфелтера
Иногда эффективно раннее лечение мужскими половыми гормонами.
XXY и XXXY- синдром Клайнфелтера
Иногда эффективно раннее лечение мужскими половыми гормонами.

Болезнь Помпе
:
Болезнь Помпе
:

Болезнь Помпе
Прогрессирующее полисистемное , инвалидизирующее, нередко, фатальное нервно-мышечное заболевание
Болезнь накопления гликогена
Болезнь Помпе
Прогрессирующее полисистемное , инвалидизирующее, нередко, фатальное нервно-мышечное заболевание
Болезнь накопления гликогена
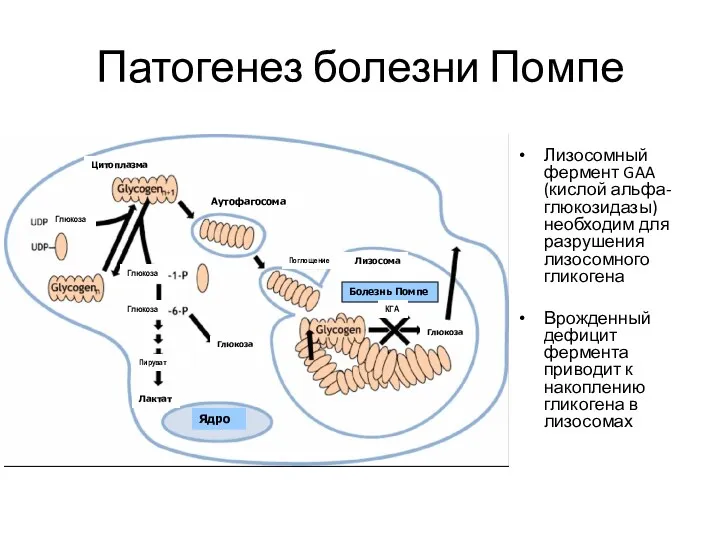
Патогенез болезни Помпе
Лизосомный фермент GAA (кислой альфа-глюкозидазы) необходим для разрушения лизосомного
Патогенез болезни Помпе
Лизосомный фермент GAA (кислой альфа-глюкозидазы) необходим для разрушения лизосомного

Генетика и частота
Болезнь Помпе моногенное
заболевание
Ген GAA локализован в длинном
Генетика и частота
Болезнь Помпе моногенное
заболевание
Ген GAA локализован в длинном

Болезнь Помпе
Характеризуется дефицитом лизосомального фермента кислой альфа-мальтазы (глюкозидазы) (GAA)
Как следствие, происходит
Болезнь Помпе
Характеризуется дефицитом лизосомального фермента кислой альфа-мальтазы (глюкозидазы) (GAA)
Как следствие, происходит

Различная скорость прогрессирования болезни
Течение болезни Помпе
Быстро прогрессирующее
Медленно прогрессирующее
Различная скорость прогрессирования болезни
Течение болезни Помпе
Быстро прогрессирующее
Медленно прогрессирующее

Скелетные мышцы
Глубокая и быстро прогрессирующая мышечная слабость
Мышечная гипотония
Амиотония
Запрокидывание головы
Значительно повышенная сывороточная
Скелетные мышцы
Глубокая и быстро прогрессирующая мышечная слабость
Мышечная гипотония
Амиотония
Запрокидывание головы
Значительно повышенная сывороточная

Инфантильная форма
Инфантильная форма

Ювенильная форма
Ювенильная форма

БОЛЕЗНЬ ПОМПЕ У ДЕТЕЙ И ВЗРОСЛЫХ
Дыхательная система
Дыхательная недостаточность/дистресс синдром
Слабость диафрагмы
Нарушения
БОЛЕЗНЬ ПОМПЕ У ДЕТЕЙ И ВЗРОСЛЫХ
Дыхательная система
Дыхательная недостаточность/дистресс синдром
Слабость диафрагмы
Нарушения

Сердце
Рентгенограмма грудной клетки
Электрокардиография (ЭКГ)
Эхокардиография (Эхо-КГ)
Легкие
Спирометрия (оценка ЖЕЛ сидя, лежа)
Рентгенограмма грудной
Сердце
Рентгенограмма грудной клетки
Электрокардиография (ЭКГ)
Эхокардиография (Эхо-КГ)
Легкие
Спирометрия (оценка ЖЕЛ сидя, лежа)
Рентгенограмма грудной

Мышцы
Электромиография (ЭМГ)/исследования нервной проводимости
Тестирование мышечной силы
Лабораторные исследования
Сывороточная креатинкиназа (КК)
Аланиновая и
Мышцы
Электромиография (ЭМГ)/исследования нервной проводимости
Тестирование мышечной силы
Лабораторные исследования
Сывороточная креатинкиназа (КК)
Аланиновая и

ДИАГНОСТИКА
Инвазивные методы:
биопсия кожи
биопсия мышцы
В настоящее время анализ сухих пятен крови
ДИАГНОСТИКА
Инвазивные методы:
биопсия кожи
биопсия мышцы
В настоящее время анализ сухих пятен крови

ЭЛЕКТРОННАЯ МИКРОСКОПИЯ
Здоровые миофибриллы замещаются гликогеном, что постепенно нарушает мышечную функцию
Изменения структуры
ЭЛЕКТРОННАЯ МИКРОСКОПИЯ
Здоровые миофибриллы замещаются гликогеном, что постепенно нарушает мышечную функцию
Изменения структуры
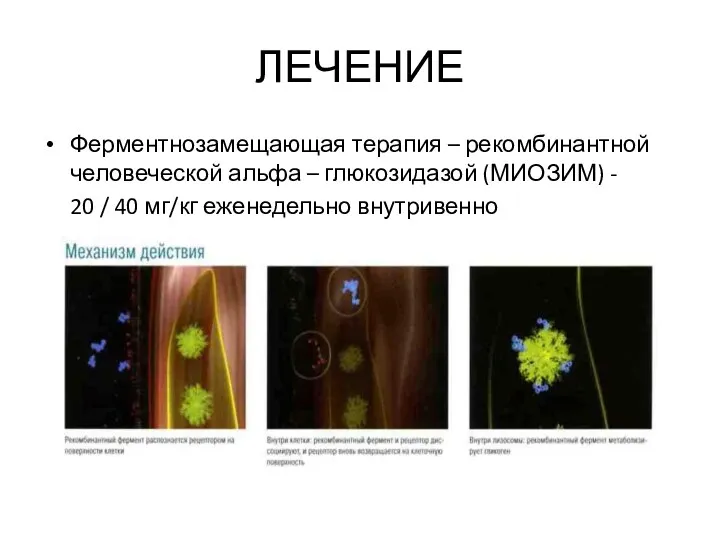
ЛЕЧЕНИЕ
Ферментнозамещающая терапия – рекомбинантной человеческой альфа – глюкозидазой (МИОЗИМ) -
20
ЛЕЧЕНИЕ
Ферментнозамещающая терапия – рекомбинантной человеческой альфа – глюкозидазой (МИОЗИМ) -
20

Результаты лечения
Результаты лечения

РЕЗУЛЬТАТЫ ЛЕЧЕНИЯ
Уменьшение потребности в ИВЛ
Увеличение ЖЕЛ, возрастание мобильности и способности передвигаться
Обретение
РЕЗУЛЬТАТЫ ЛЕЧЕНИЯ
Уменьшение потребности в ИВЛ
Увеличение ЖЕЛ, возрастание мобильности и способности передвигаться
Обретение
БОЛЕЗНЬ ФАБРИ
БОЛЕЗНЬ ФАБРИ

Болезнь Фабри
Наследственный дефект фермента α-галактозидазы А, приводящий к прогрессерующему накоплению
Болезнь Фабри
Наследственный дефект фермента α-галактозидазы А, приводящий к прогрессерующему накоплению

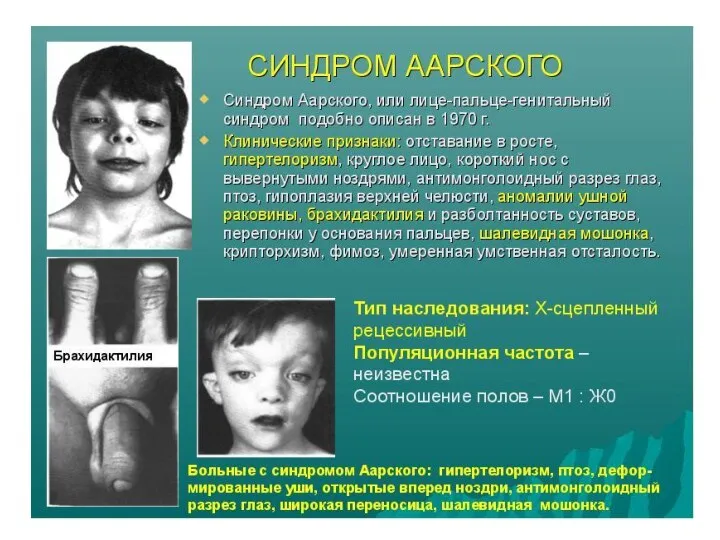
Фенотип пациентов с БФ
Фенотип пациентов с БФ

ОСНОВНЫЕ ПРИЗНАКИ
Увеличены нос, уши, язык, слюнные железы
Диспропорциональный рост костей черепа (увеличение
ОСНОВНЫЕ ПРИЗНАКИ
Увеличены нос, уши, язык, слюнные железы
Диспропорциональный рост костей черепа (увеличение

Тип наследования–
Х-сцепленный рецессивный
Тип наследования–
Х-сцепленный рецессивный
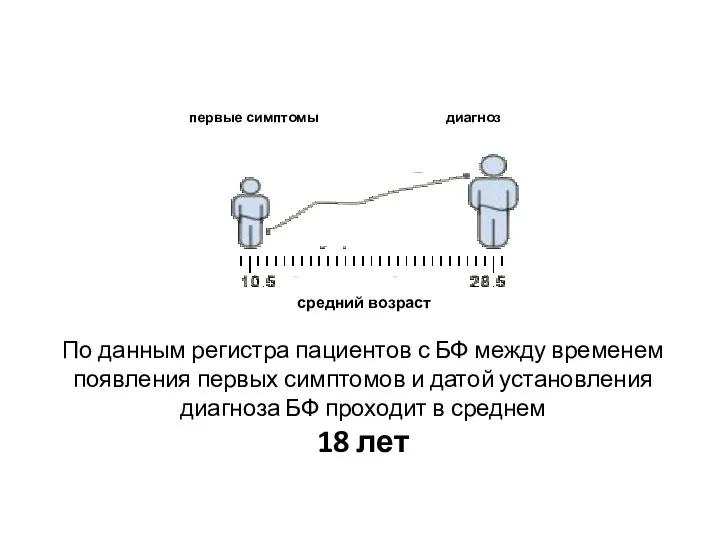
По данным регистра пациентов с БФ между временем появления первых симптомов
По данным регистра пациентов с БФ между временем появления первых симптомов
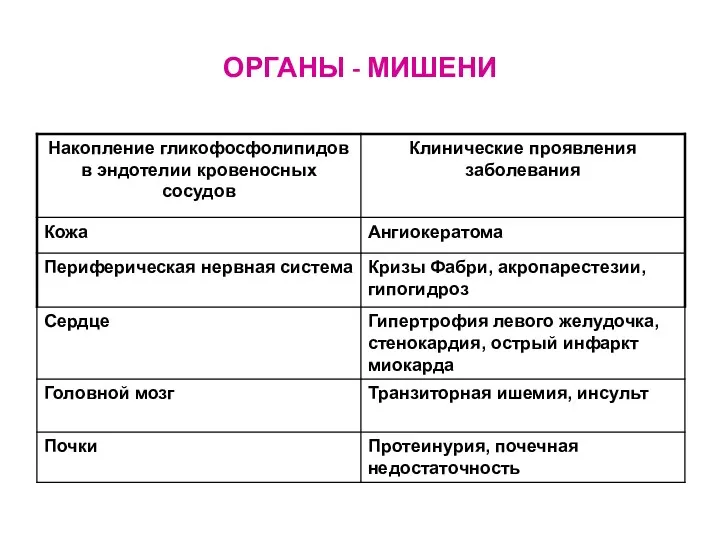
ОРГАНЫ - МИШЕНИ
ОРГАНЫ - МИШЕНИ
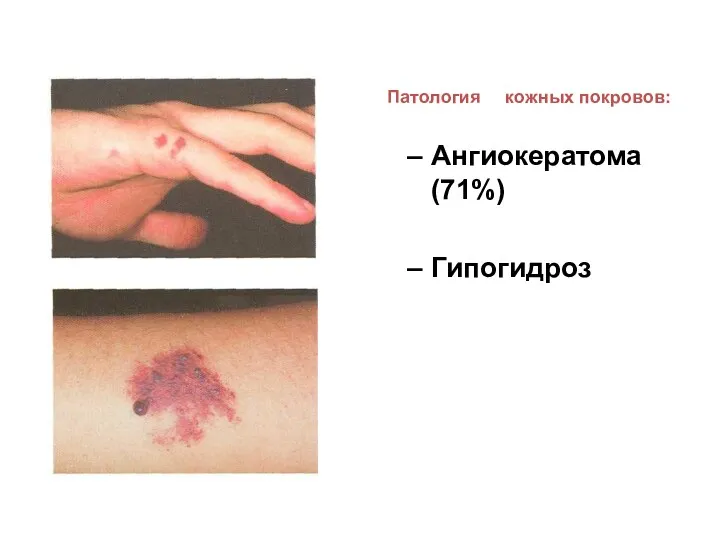
Патология кожных покровов:
Ангиокератома (71%)
Гипогидроз
Патология кожных покровов:
Ангиокератома (71%)
Гипогидроз
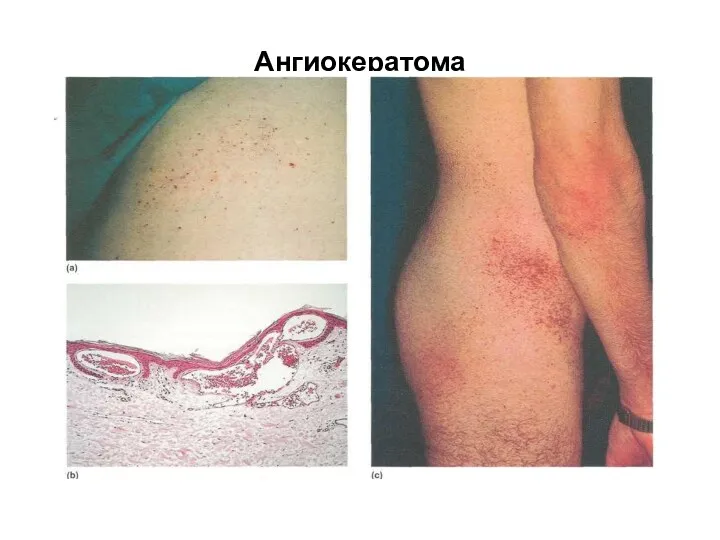
Ангиокератома
Ангиокератома

Патология периферической нервной системы
кризы Фабри –жгучие боли в ладонях и
Патология периферической нервной системы
кризы Фабри –жгучие боли в ладонях и
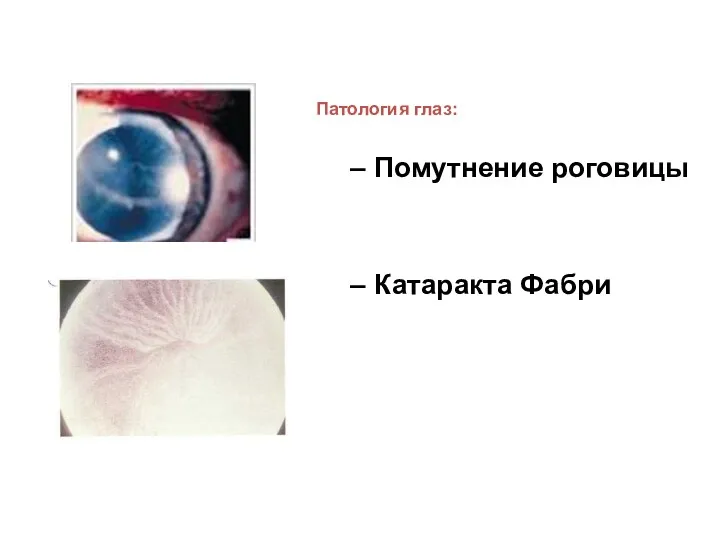
Патология глаз:
Помутнение роговицы
Катаракта Фабри
Патология глаз:
Помутнение роговицы
Катаракта Фабри

Патология глаз:
Извитость и аневризмы сосудов сетчатки
Ретинопатия
слепота
Патология глаз:
Извитость и аневризмы сосудов сетчатки
Ретинопатия
слепота
Патология сердца:
Гипертрофия левого желудочка (88%)
Недостаточность митрального клапана
Нарушения ритма и проводимости
Стенокардия
Инфаркт
Патология сердца:
Гипертрофия левого желудочка (88%)
Недостаточность митрального клапана
Нарушения ритма и проводимости
Стенокардия
Инфаркт

Цереброваскулярная недостаточность
Транзиторные ишемические атаки (гемиплегия, гемианестезия, афазия, судороги)
Инсульты с характерными изменениями
Цереброваскулярная недостаточность
Транзиторные ишемические атаки (гемиплегия, гемианестезия, афазия, судороги)
Инсульты с характерными изменениями
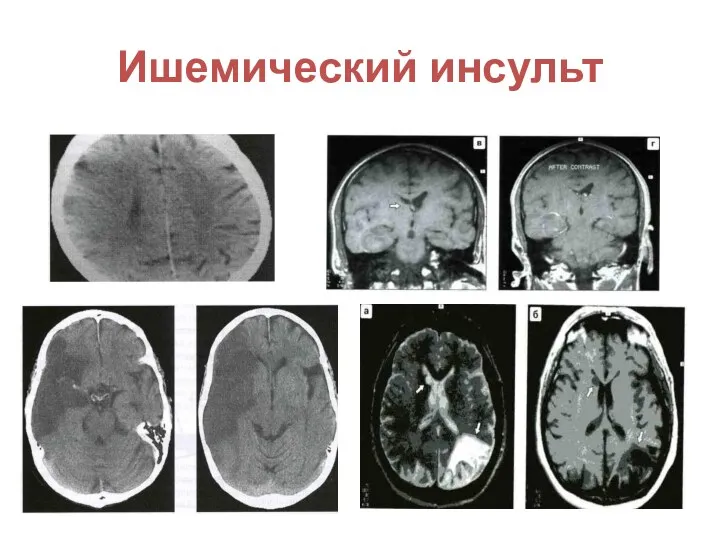
Ишемический инсульт
Ишемический инсульт

Патология почек:
Протеинурия (84%)
Тубулярная дисфункция
Высокий уровень креатинина
Почечная недостаточность (47%)
Патология почек:
Протеинурия (84%)
Тубулярная дисфункция
Высокий уровень креатинина
Почечная недостаточность (47%)

Неспецифические проявления БФ
Нейросенсорная тугоухость
Хронические обструктивные заболевания бронхов
Патология ЖКТ: тошнота, рвота, абдоминальные
Неспецифические проявления БФ
Нейросенсорная тугоухость
Хронические обструктивные заболевания бронхов
Патология ЖКТ: тошнота, рвота, абдоминальные

Лабораторные данные
Анализ мочи:протеинурия, гематурия, цилиндрурия, эпителий почечных канальцев, липидные глобулы в
Лабораторные данные
Анализ мочи:протеинурия, гематурия, цилиндрурия, эпителий почечных канальцев, липидные глобулы в

Традиционная диагностика ЛБН
Измерение активности ферментов в:
Лейкоцитах, изолированных из крови с ЭДТА
Лимфоцитах,
Традиционная диагностика ЛБН
Измерение активности ферментов в:
Лейкоцитах, изолированных из крови с ЭДТА
Лимфоцитах,

Преимущества метода DBS
Позволяет осуществлять пересылку образцов крови
Небольшой объем крови для исследования
Высушенные
Преимущества метода DBS
Позволяет осуществлять пересылку образцов крови
Небольшой объем крови для исследования
Высушенные

Патогенетическая терапия при болезни Фабри
Трансплантация стволовых кроветворных клеток
Трансплантация печени плода
Геннотерапия
Энзимо-заместительная терапия
Патогенетическая терапия при болезни Фабри
Трансплантация стволовых кроветворных клеток
Трансплантация печени плода
Геннотерапия
Энзимо-заместительная терапия

Фабразим (агалзидаза бета) – человеческая рекомбинантная альфа-галактозидаза А полностью идентичная нативному
Фабразим (агалзидаза бета) – человеческая рекомбинантная альфа-галактозидаза А полностью идентичная нативному

Фабразим (агалзидаза бета)
Флаконы – 35мг и 5 мг
Назначается в дозе 1
Фабразим (агалзидаза бета)
Флаконы – 35мг и 5 мг
Назначается в дозе 1
 презентация на тему Закрепляем правила дорожного движения.
презентация на тему Закрепляем правила дорожного движения. Информационные технологии производственного менеджмента на предприятии
Информационные технологии производственного менеджмента на предприятии Проектирование энергоэффективных зданий
Проектирование энергоэффективных зданий Литературные места России. Тамань
Литературные места России. Тамань Игра-презентация Веселое путешествие
Игра-презентация Веселое путешествие Презентация Советы для подготовки к ЕГЭ
Презентация Советы для подготовки к ЕГЭ Влияние мультфильмов на развитие ребенка дошкольного возраста
Влияние мультфильмов на развитие ребенка дошкольного возраста Объективные предпосылки международной экономической интеграции
Объективные предпосылки международной экономической интеграции Методическая разработка урока по теме Фосфор Тема урока: Фосфор
Методическая разработка урока по теме Фосфор Тема урока: Фосфор Рок 21 века
Рок 21 века Проектирование программы духовно-нравственного воспитания
Проектирование программы духовно-нравственного воспитания 10 новых украинских заводов
10 новых украинских заводов Близость и отделение – основные механизмы развития семьи
Близость и отделение – основные механизмы развития семьи Генераторы звукового диапазона частот
Генераторы звукового диапазона частот Транзактный анализ Э. Берна. Сценарий жизни
Транзактный анализ Э. Берна. Сценарий жизни Обогащение полезных ископаемых. Процессы и аппараты обогащения, химическое обогащение, комбинированные технологии
Обогащение полезных ископаемых. Процессы и аппараты обогащения, химическое обогащение, комбинированные технологии Разработка первичной модели и конструкции женского платья для повседневной носки. Рукав втачной. Силуэт прилегающий
Разработка первичной модели и конструкции женского платья для повседневной носки. Рукав втачной. Силуэт прилегающий АРМ журналиста/ писателя. Архитектура компьютера
АРМ журналиста/ писателя. Архитектура компьютера Презентация без названия (wecompress.com)
Презентация без названия (wecompress.com) Let’s face it, it’s hard to describe
Let’s face it, it’s hard to describe Неотложные состояния в оториноларингологии
Неотложные состояния в оториноларингологии Наглядное пособие
Наглядное пособие Рисуем Медузу
Рисуем Медузу презентация Турнир Почемучек
презентация Турнир Почемучек Несостоятельность рынка и государства
Несостоятельность рынка и государства Семья в современном обществе и ее функции. Влияние семейных отношений на здоровье человека
Семья в современном обществе и ее функции. Влияние семейных отношений на здоровье человека Научная работа с презентацией Давать прозвища и клички - плохая привычка
Научная работа с презентацией Давать прозвища и клички - плохая привычка Robert Burns (1759–1796)
Robert Burns (1759–1796)