- Наследственные болезни энзимопатии

Содержание
- 2. План Энзимопатии Причины возникновений наследственных энзимопатий Энзимопатии обмена аминокислот Энзимопатии обмена углеводов Энзимопатии обмена липидов Энзимопатии
- 3. Энзимопатии (ферментопатии) в широком смысле слова — патологические изменения активности ферментов. В более узком смысле этим
- 4. Алиментарная (приобретенная) Ферментопатии Вследствие мутации При хроническом расстройстве питания (белковое голодание) Наследственная
- 5. Наследственные болезни —заболевания человека, обусловленные хромосомными и генными мутациями Наследственные энзимопатии связаны с генетически обусловленной недостаточностью
- 6. Причины возникновения энзимопатий Полная блокада (выключение) синтеза фермента; Снижения активности фермента; Нарушения других систем или биохимических
- 7. Особенностью течения наследственных энзимопатий является наличие скрытого периода, когда болезнь не имеет выраженных клин. симптомов, но
- 8. Наследственные болезни обмена аминокислот(ФКУ) фенилкетонурия характеризуется нарушениями функции центральной нервной системы, что проявляется изменением мышечного тонуса,
- 9. Фенилкетонурия Впервые описал A. Foiling в 1934 году. Поражение ЦНС вызывается недостаточностью фермента гидроксилазы-4-фенилаланина, управляющего превращением
- 10. Альбинизм. Блокада активности фермента тирозиназы, катализирующей синтез меланина из тирозина через дигидроксифенилаланин Основными проявлениями ее служат
- 12. Лечение 1 Ограничение в диете белка и соответствующей аминокислоты. 2. Дополнительное назначение незаменимых аминокислот. 3. Назначение
- 13. К наследственным болезням углеводного обмена относят гликогенозы, галактоземию, нек-рые формы сахарного диабета и др. Наследственные болезни
- 14. Галактоземия Описана в 1908 году, однако дефект обмена, ее обуславливающий, был открыт лишь в 1956 году.
- 15. Патогенез Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не усваивается, а промежуточный продукт
- 16. Клиника Проявляется вскоре после рождения у ребенка: отказом от пищи, поносом, рвотой, непереносимостью голода, падением массы
- 17. Наследственные болезни пуринового и пиримидинового обмена включают некоторые формы подагры, синдром Леша-Найхана (возникает при недостаточности гипоксантин
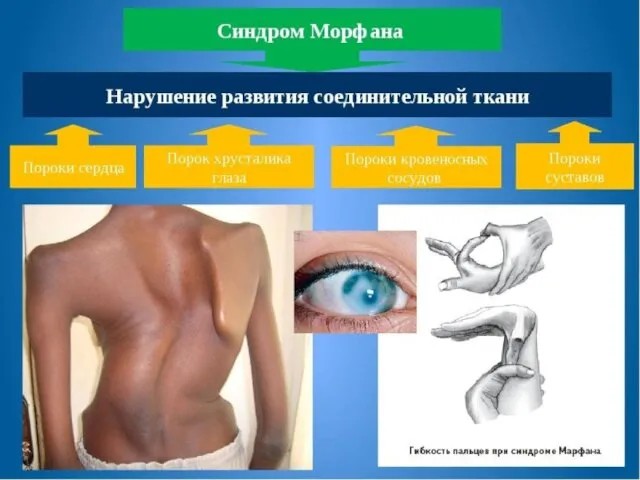
- 18. Наследственными болезнями обмена соединительной ткани Являются мукополисахаридозы, синдром Марфана, хондродистрофия; Наследственными болезнями крови и кроветворных органов
- 20. МУКОПОЛИСАХАРИДО3 (СИНДРОМ ГУРЛЕРА) Описан G. Gurler в 1919 году. Встречается с частотой — 1: 40 000.
- 21. Клиника Проявляется на первом году жизни. Внешний вид больных — увеличенная голова, выдающиеся лобные бугры, почти
- 22. Психическое состояние Умственная отсталость заметна уже в раннем возрасте. В последующем интеллектуальный дефект усугубляется, затем происходит
- 23. Основное значение в диагностике наследственных энзимопатий имеют биохимические методы исследования: определение активности ферментов, продуктов обмена веществ,
- 25. Скачать презентацию

План
Энзимопатии
Причины возникновений наследственных энзимопатий
Энзимопатии обмена аминокислот
Энзимопатии обмена углеводов
План
Энзимопатии
Причины возникновений наследственных энзимопатий
Энзимопатии обмена аминокислот
Энзимопатии обмена углеводов

Энзимопатии (ферментопатии) в широком смысле слова — патологические изменения активности ферментов.
Энзимопатии (ферментопатии) в широком смысле слова — патологические изменения активности ферментов.
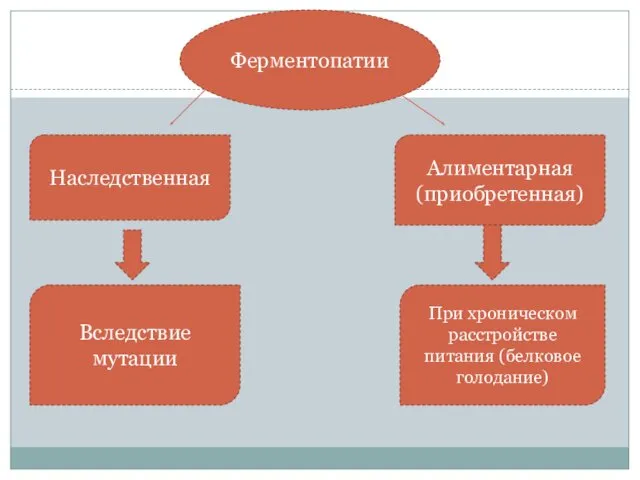
Алиментарная
(приобретенная)
Ферментопатии
Вследствие мутации
При хроническом расстройстве питания (белковое голодание)
Наследственная
Алиментарная
(приобретенная)
Ферментопатии
Вследствие мутации
При хроническом расстройстве питания (белковое голодание)
Наследственная

Наследственные болезни —заболевания человека, обусловленные хромосомными и генными мутациями
Наследственные энзимопатии связаны
Наследственные болезни —заболевания человека, обусловленные хромосомными и генными мутациями
Наследственные энзимопатии связаны

Причины возникновения энзимопатий
Полная блокада (выключение) синтеза фермента;
Снижения активности фермента;
Нарушения
Причины возникновения энзимопатий
Полная блокада (выключение) синтеза фермента;
Снижения активности фермента;
Нарушения

Особенностью течения наследственных энзимопатий
является наличие скрытого периода, когда болезнь не имеет
Особенностью течения наследственных энзимопатий
является наличие скрытого периода, когда болезнь не имеет

Наследственные болезни обмена аминокислот(ФКУ)
фенилкетонурия характеризуется нарушениями функции центральной нервной системы, что
Наследственные болезни обмена аминокислот(ФКУ)
фенилкетонурия характеризуется нарушениями функции центральной нервной системы, что

Фенилкетонурия
Впервые описал A. Foiling в 1934 году.
Поражение ЦНС вызывается недостаточностью
Фенилкетонурия
Впервые описал A. Foiling в 1934 году.
Поражение ЦНС вызывается недостаточностью

Альбинизм.
Блокада активности фермента тирозиназы, катализирующей синтез меланина из тирозина через дигидроксифенилаланин
Основными
Альбинизм.
Блокада активности фермента тирозиназы, катализирующей синтез меланина из тирозина через дигидроксифенилаланин
Основными


Лечение
1 Ограничение в диете белка и соответствующей аминокислоты.
2. Дополнительное назначение незаменимых
Лечение
1 Ограничение в диете белка и соответствующей аминокислоты.
2. Дополнительное назначение незаменимых

К наследственным болезням углеводного обмена
относят гликогенозы, галактоземию, нек-рые формы сахарного диабета
К наследственным болезням углеводного обмена
относят гликогенозы, галактоземию, нек-рые формы сахарного диабета
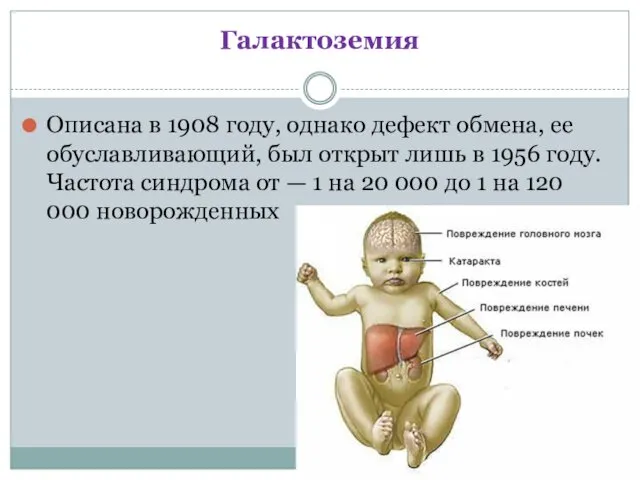
Галактоземия
Описана в 1908 году, однако дефект обмена, ее обуславливающий, был открыт
Галактоземия
Описана в 1908 году, однако дефект обмена, ее обуславливающий, был открыт

Патогенез
Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не
Патогенез
Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не

Клиника
Проявляется вскоре после рождения у ребенка: отказом от пищи, поносом, рвотой,
Клиника
Проявляется вскоре после рождения у ребенка: отказом от пищи, поносом, рвотой,

Наследственные болезни пуринового и пиримидинового обмена
включают некоторые формы подагры, синдром Леша-Найхана
Наследственные болезни пуринового и пиримидинового обмена
включают некоторые формы подагры, синдром Леша-Найхана

Наследственными болезнями обмена соединительной ткани
Являются мукополисахаридозы, синдром Марфана, хондродистрофия;
Наследственными болезнями крови
Наследственными болезнями обмена соединительной ткани
Являются мукополисахаридозы, синдром Марфана, хондродистрофия;
Наследственными болезнями крови
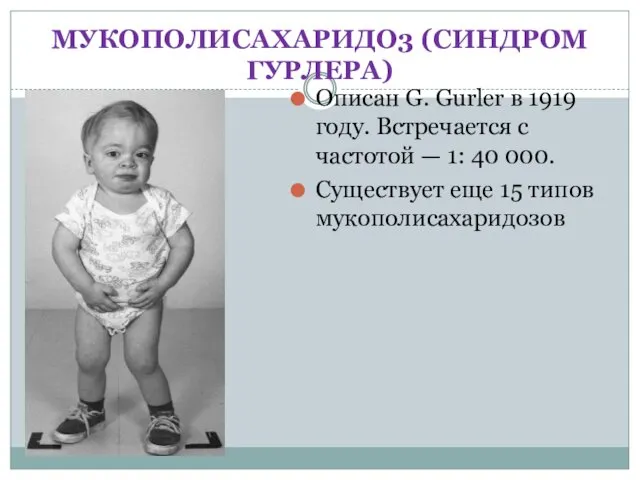
МУКОПОЛИСАХАРИДО3 (СИНДРОМ ГУРЛЕРА)
Описан G. Gurler в 1919 году. Встречается с частотой
МУКОПОЛИСАХАРИДО3 (СИНДРОМ ГУРЛЕРА)
Описан G. Gurler в 1919 году. Встречается с частотой

Клиника
Проявляется на первом году жизни.
Внешний вид больных — увеличенная голова,
Клиника
Проявляется на первом году жизни.
Внешний вид больных — увеличенная голова,

Психическое состояние
Умственная отсталость заметна уже в раннем возрасте. В последующем интеллектуальный
Психическое состояние
Умственная отсталость заметна уже в раннем возрасте. В последующем интеллектуальный

Основное значение в диагностике наследственных энзимопатий имеют биохимические методы исследования: определение
Основное значение в диагностике наследственных энзимопатий имеют биохимические методы исследования: определение
 Пищеварительная система
Пищеварительная система Шаблон 9 мая
Шаблон 9 мая Сферы применения роботов
Сферы применения роботов Поняття про полімери на прикладі поліетилену. Використання поліетилену. 9 клас
Поняття про полімери на прикладі поліетилену. Використання поліетилену. 9 клас Убинский сельсовет Убинского района Новосибирской области. Формирование комфортной городской среды
Убинский сельсовет Убинского района Новосибирской области. Формирование комфортной городской среды 20190508_informatsiya._kolichestvo_informatsii
20190508_informatsiya._kolichestvo_informatsii Моя картина мира
Моя картина мира Лекарственные растения Нижегородской области
Лекарственные растения Нижегородской области Оборудование игровой комнаты для детей от 1,5 до 3 лет
Оборудование игровой комнаты для детей от 1,5 до 3 лет Откровенье вечной красоты
Откровенье вечной красоты Аргентина
Аргентина Пастель - (фр. pastel, от лат. pasta — тесто) сухая живопись мягкими цветными мелками
Пастель - (фр. pastel, от лат. pasta — тесто) сухая живопись мягкими цветными мелками Роман Л.Н.Толстого Война и мир: история создания, своеобразие
Роман Л.Н.Толстого Война и мир: история создания, своеобразие Нетрадиционное рисование
Нетрадиционное рисование Перпендикуляр и наклонная
Перпендикуляр и наклонная Презентация Формирование творческих компетенций детей в процессе рисования нетрадиционными техниками и материалами
Презентация Формирование творческих компетенций детей в процессе рисования нетрадиционными техниками и материалами Моделирование и конструирование одежды промышленного производства
Моделирование и конструирование одежды промышленного производства Презентация Рука развивает мозг
Презентация Рука развивает мозг Я и моя семья, Сергеев Костя
Я и моя семья, Сергеев Костя Электромагнитное поле. (Лекция 5)
Электромагнитное поле. (Лекция 5) Четырехволновое смешение света
Четырехволновое смешение света Задачи в стихах
Задачи в стихах Modal Verbs
Modal Verbs Проект cоциально-экономического развития территорий Тульской области на основе родовых поместий
Проект cоциально-экономического развития территорий Тульской области на основе родовых поместий Страны Зарубежной Азии
Страны Зарубежной Азии Основы технологии оклейки стен обоями
Основы технологии оклейки стен обоями Цифровые технологии печати
Цифровые технологии печати Выступление Современный урок в условиях реализации национальной образовательной инициативы Наша новая Школа
Выступление Современный урок в условиях реализации национальной образовательной инициативы Наша новая Школа