- Редкие болезни в детском возрасте (Орфанные болезни)

Содержание
- 2. Определение Орфанные заболевания (англ. rare disease, orphan disease) — заболевания, затрагивающие небольшую часть популяции. Орфанные болезни
- 3. Распространенность Распространенность орфанных болезней составляет около 1 : 2 000 и реже по рекомендации European Commission
- 4. Определение В разных странах определение и перечень орфанных болезней принимаются на государственном уровне. Нет единого определения
- 5. Актуальность проблемы В настоящее время в развитых странах ведется активное изучение орфанных заболеваний. Оно затрудняется малым
- 6. Особенности орфанных заболеваний Примерно половина орфанных заболеваний обусловлена генетическими отклонениями. Симптомы могут быть очевидны с рождения
- 7. Особенности орфанных заболеваний Большинство орфанных заболеваний – хронические. Они в значительной мере ухудшают качество жизни человека
- 8. Эпидемиология В России редкими предложено считать заболевания с «распространенностью не более 10 случаев на 100 000
- 9. Пациенты 24-х орфанных болезней обеспечиваются лекарственными препаратами и лечебным питанием за счет государства ( Постановление правительства
- 10. Неонатальный скрининг В рамках национального проекта «Здоровье» все новорожденные проходят диагностику (неонатальный скрининг) на 5 наследственных
- 11. Перечень редких заболеваний в России Во исполнение статьи 44 Федерального закона от 21 ноября 2011 г.
- 12. Перечень редких заболеваний в России Метахроматическая лейкодистрофия (арилсульфатаза) ( 22q13) Лейкодистрофия Краббе Х-сцепленная адренолейкодистрофия Витамин Д-резистентный
- 13. Перечень редких заболеваний в России Болезнь Фабри Гистидинемия Муковисцидоз Гомоцистинурия( нарушение метаболизма кобаламина) 3-метилглутаконовая ацидемия Несовершенное
- 14. Перечень редких заболеваний в России Цистиноз Гиперлизинемия Мукополисахаридоз III тип Мукополисахаридоз IV тип Мукополисахаридоз VII тип
- 15. Перечень редких заболеваний в России Синдром Стиклера Синдром Билса Синдром Вейла-Марчезани Нейрофиброматоз I типа Туберозный склероз
- 16. Перечень редких заболеваний в России Дефицит ацетил-коэнзим А дегидрогеназы жирных кислот с короткой углеродной цепью; с
- 17. Мукополисахаридозы Группа наследственных болезней соединительной ткани с генетически обусловленной неполноценностью 1 из 11 известных ферментов, участвующих
- 18. Для МПС характерно полисистемное поражение: множественные деформации скелета, задержка физического развития, умственная отсталость при некоторых формах,
- 19. Hunter синдром Тип наследования- аутосомио-рецессивный, за исключением МПС II типа (болезнь Хантера наследуется по X-сцепленному рецессивному
- 20. Клинические проявления (Hunter синдром) Задержка умственного развития; Гепатоспленомегалия Множественные дизостозы: утолщение кости кистей и черепа, деформация
- 21. Мукополисахаридоз II типа (Hunter) Мукополисахаридозы включены в группу лизосомальных болезней «накопления». Мукополисахариды составляют основу соединительной ткани.
- 22. Hunter синдром Дифференциальный диагноз проводится внутри группы МПС и с другими лизосомными болезнями накопления: муколипидозы, ганглиозидозы,
- 23. Диагностика 1. Определение уровня экскреции ГАГ ( глюкозоамингликанов) с мочой (возрастает при заболевании), 2. Измерение активности
- 24. ЛЕЧЕНИЕ (Hunter синдром) Лечение : проводится пожизненная заместительная ферментотерапия . При МПС ll типа используют препарат
- 25. Лейкодистрофии Лейкодистрофии - это группа заболеваний, характеризующихся прогрессирующим поражением белого вещества головного мозга и глии. Тип
- 26. Формы лейкодистрофии Первый вариант (тип) - встречается довольно редко и называется врожденным; клиническая картина разворачивается в
- 27. Формы лейкодистрофии Второй вариант (тип) - наиболее распространенный, его называют ювенильным. Симптомы при этой форме заболевания
- 28. Формы лейкодистрофии Взрослая форма начинается на втором-третьем десятилетии жизни. Клинические проявления заболевания характеризуются наличием выраженных психических
- 29. Лейкодистрофия II типа Клинический случай Больной Б., 5,5 лет. Впервые обратился в возрасте 2 лет с
- 30. Лейкодистрофия II типа Клинический случай К 3-му месяцу жизни ребенок самостоятельно не удерживал голову, отмечались усиление
- 31. Лейкодистрофия II типа Клинический случай В 8 мес: неврологический статус оставался прежним, однако отмечалось нарастание мышечного
- 32. Данные объективного исследования Осмотр в 5,5 лет: Форма головы — гидроцефальная с выраженными лобно-теменными буграми. Множественные
- 33. Данные объективного исследования Высшие когнитивные функции: грубое нарушение психического развития — мало интересуется окружающим, однако узнает
- 34. Данные дополнительных методов исследования Компьютерная ЭЭГ: грубые огранические изменения биоэлектрической активности, ирритативные общемозговые изменения биоэлектрической активности.
- 35. Данные дополнительных методов исследования KT головного мозга: на серии сагиттальных томограмм (6 мес, 1 год 1
- 36. Заключение по больному Большие трудности представляет клиническая диагностика нейродегенеративных заболеваний ввиду их редкости, а также неспецифичности
- 37. Муковисцидоз Муковисцидоз встречается у людей белой расы, распостраненность в Европе - 1:2800-1:9800. Одинаково склонны к болезни
- 38. Патогенез Мукосекреторные железы выделяют вязкую слизь, (железы слизистой рта, пищевода, кишечника, поджелудочной железы, носа, синусов, трахеи,
- 39. Классификация 1. Смешанная форма (поражение дыхательной и пищеварительной систем). 2. Легочная форма. 3. Кишечная форма. 4.
- 40. Диагностика Диагностика заболевания основывается на симптомокомплексе, который состоит из врожденной гипотрофии, поздней и медленной эвакуации густого
- 41. Лечение Лечение направлено на восстановление дыхательной и пищеварительной функций. Прогноз зависит от тяжести заболевания. Если диагноз
- 42. Пример успешного лечения кишечной формы муковисцидоза. На фотографиях пациент в период диагностики до начала терапии и
- 44. Скачать презентацию

Определение
Орфанные заболевания (англ. rare disease, orphan disease) —
заболевания, затрагивающие небольшую
Определение
Орфанные заболевания (англ. rare disease, orphan disease) —
заболевания, затрагивающие небольшую

Распространенность
Распространенность орфанных болезней составляет около 1 : 2 000 и реже
Распространенность
Распространенность орфанных болезней составляет около 1 : 2 000 и реже

Определение
В разных странах определение и перечень орфанных болезней принимаются на государственном
Определение
В разных странах определение и перечень орфанных болезней принимаются на государственном

Актуальность проблемы
В настоящее время в развитых странах ведется активное изучение орфанных
Актуальность проблемы
В настоящее время в развитых странах ведется активное изучение орфанных

Особенности орфанных заболеваний
Примерно половина орфанных заболеваний обусловлена генетическими отклонениями. Симптомы могут
Особенности орфанных заболеваний
Примерно половина орфанных заболеваний обусловлена генетическими отклонениями. Симптомы могут

Особенности орфанных заболеваний
Большинство орфанных заболеваний – хронические. Они в значительной мере
Особенности орфанных заболеваний
Большинство орфанных заболеваний – хронические. Они в значительной мере

Эпидемиология
В России редкими предложено считать заболевания с «распространенностью не более 10
Эпидемиология
В России редкими предложено считать заболевания с «распространенностью не более 10

Пациенты 24-х орфанных болезней обеспечиваются лекарственными препаратами и лечебным питанием
Пациенты 24-х орфанных болезней обеспечиваются лекарственными препаратами и лечебным питанием

Неонатальный скрининг
В рамках национального проекта «Здоровье» все новорожденные проходят диагностику (неонатальный
Неонатальный скрининг
В рамках национального проекта «Здоровье» все новорожденные проходят диагностику (неонатальный

Перечень редких заболеваний в России
Во исполнение статьи 44 Федерального закона
Перечень редких заболеваний в России
Во исполнение статьи 44 Федерального закона

Перечень редких заболеваний в России
Метахроматическая лейкодистрофия (арилсульфатаза) ( 22q13)
Лейкодистрофия
Перечень редких заболеваний в России
Метахроматическая лейкодистрофия (арилсульфатаза) ( 22q13) Лейкодистрофия

Перечень редких заболеваний в России
Болезнь Фабри
Гистидинемия
Муковисцидоз
Гомоцистинурия( нарушение
Перечень редких заболеваний в России
Болезнь Фабри Гистидинемия Муковисцидоз Гомоцистинурия( нарушение

Перечень редких заболеваний в России
Цистиноз
Гиперлизинемия
Мукополисахаридоз III тип
Мукополисахаридоз
Перечень редких заболеваний в России
Цистиноз Гиперлизинемия Мукополисахаридоз III тип Мукополисахаридоз

Перечень редких заболеваний в России
Синдром Стиклера
Синдром Билса
Синдром Вейла-Марчезани
Перечень редких заболеваний в России
Синдром Стиклера Синдром Билса Синдром Вейла-Марчезани

Перечень редких заболеваний в России
Дефицит ацетил-коэнзим А дегидрогеназы жирных кислот
Перечень редких заболеваний в России
Дефицит ацетил-коэнзим А дегидрогеназы жирных кислот

Мукополисахаридозы
Группа наследственных болезней соединительной ткани с генетически обусловленной неполноценностью 1
Мукополисахаридозы
Группа наследственных болезней соединительной ткани с генетически обусловленной неполноценностью 1

Для МПС характерно полисистемное поражение: множественные деформации скелета, задержка физического развития,
Для МПС характерно полисистемное поражение: множественные деформации скелета, задержка физического развития,

Hunter синдром
Тип наследования- аутосомио-рецессивный, за исключением МПС II типа (болезнь Хантера
Hunter синдром
Тип наследования- аутосомио-рецессивный, за исключением МПС II типа (болезнь Хантера

Клинические проявления
(Hunter синдром)
Задержка умственного развития;
Гепатоспленомегалия
Множественные дизостозы: утолщение кости кистей
Клинические проявления
(Hunter синдром)
Задержка умственного развития;
Гепатоспленомегалия
Множественные дизостозы: утолщение кости кистей
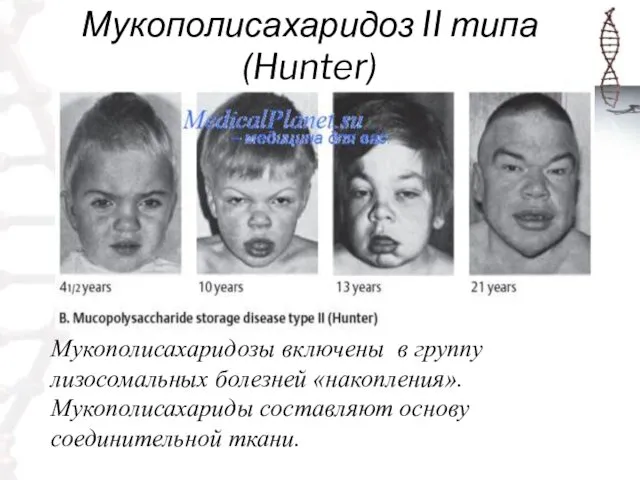
Мукополисахаридоз II типа (Hunter)
Мукополисахаридозы включены в группу лизосомальных болезней «накопления».
Мукополисахариды составляют
Мукополисахаридоз II типа (Hunter)
Мукополисахаридозы включены в группу лизосомальных болезней «накопления».
Мукополисахариды составляют

Hunter синдром
Дифференциальный диагноз проводится внутри группы МПС и с другими лизосомными
Hunter синдром
Дифференциальный диагноз проводится внутри группы МПС и с другими лизосомными

Диагностика
1. Определение уровня экскреции ГАГ ( глюкозоамингликанов) с мочой
Диагностика
1. Определение уровня экскреции ГАГ ( глюкозоамингликанов) с мочой

ЛЕЧЕНИЕ
(Hunter синдром)
Лечение : проводится пожизненная заместительная ферментотерапия . При МПС ll
ЛЕЧЕНИЕ
(Hunter синдром)
Лечение : проводится пожизненная заместительная ферментотерапия . При МПС ll

Лейкодистрофии
Лейкодистрофии - это группа заболеваний, характеризующихся прогрессирующим поражением белого вещества головного
Лейкодистрофии
Лейкодистрофии - это группа заболеваний, характеризующихся прогрессирующим поражением белого вещества головного

Формы лейкодистрофии
Первый вариант (тип) - встречается довольно редко и называется врожденным;
Формы лейкодистрофии
Первый вариант (тип) - встречается довольно редко и называется врожденным;

Формы лейкодистрофии
Второй вариант (тип) - наиболее распространенный, его называют ювенильным. Симптомы
Формы лейкодистрофии
Второй вариант (тип) - наиболее распространенный, его называют ювенильным. Симптомы

Формы лейкодистрофии
Взрослая форма начинается на втором-третьем десятилетии жизни. Клинические проявления заболевания
Формы лейкодистрофии
Взрослая форма начинается на втором-третьем десятилетии жизни. Клинические проявления заболевания

Лейкодистрофия II типа
Клинический случай
Больной Б., 5,5 лет. Впервые обратился в возрасте
Лейкодистрофия II типа
Клинический случай
Больной Б., 5,5 лет. Впервые обратился в возрасте

Лейкодистрофия II типа
Клинический случай
К 3-му месяцу жизни ребенок самостоятельно не удерживал
Лейкодистрофия II типа
Клинический случай
К 3-му месяцу жизни ребенок самостоятельно не удерживал

Лейкодистрофия II типа
Клинический случай
В 8 мес: неврологический статус оставался прежним, однако
Лейкодистрофия II типа
Клинический случай
В 8 мес: неврологический статус оставался прежним, однако

Данные объективного исследования
Осмотр в 5,5 лет:
Форма головы — гидроцефальная с выраженными
Данные объективного исследования
Осмотр в 5,5 лет:
Форма головы — гидроцефальная с выраженными

Данные объективного исследования
Высшие когнитивные функции: грубое нарушение психического развития — мало
Данные объективного исследования
Высшие когнитивные функции: грубое нарушение психического развития — мало

Данные дополнительных методов исследования
Компьютерная ЭЭГ: грубые огранические изменения биоэлектрической активности, ирритативные
Данные дополнительных методов исследования
Компьютерная ЭЭГ: грубые огранические изменения биоэлектрической активности, ирритативные

Данные дополнительных методов исследования
KT головного мозга: на серии сагиттальных томограмм (6
Данные дополнительных методов исследования
KT головного мозга: на серии сагиттальных томограмм (6

Заключение по больному
Большие трудности представляет клиническая диагностика нейродегенеративных заболеваний ввиду их
Заключение по больному
Большие трудности представляет клиническая диагностика нейродегенеративных заболеваний ввиду их

Муковисцидоз
Муковисцидоз встречается у людей белой расы, распостраненность в Европе - 1:2800-1:9800.
Муковисцидоз
Муковисцидоз встречается у людей белой расы, распостраненность в Европе - 1:2800-1:9800.

Патогенез
Мукосекреторные железы выделяют вязкую слизь, (железы слизистой рта, пищевода, кишечника, поджелудочной
Патогенез
Мукосекреторные железы выделяют вязкую слизь, (железы слизистой рта, пищевода, кишечника, поджелудочной

Классификация
1. Смешанная форма (поражение дыхательной и пищеварительной систем).
2. Легочная
Классификация
1. Смешанная форма (поражение дыхательной и пищеварительной систем). 2. Легочная

Диагностика
Диагностика заболевания основывается на симптомокомплексе, который состоит из врожденной гипотрофии, поздней
Диагностика
Диагностика заболевания основывается на симптомокомплексе, который состоит из врожденной гипотрофии, поздней

Лечение
Лечение направлено на восстановление дыхательной и пищеварительной функций.
Прогноз зависит от тяжести
Лечение
Лечение направлено на восстановление дыхательной и пищеварительной функций.
Прогноз зависит от тяжести
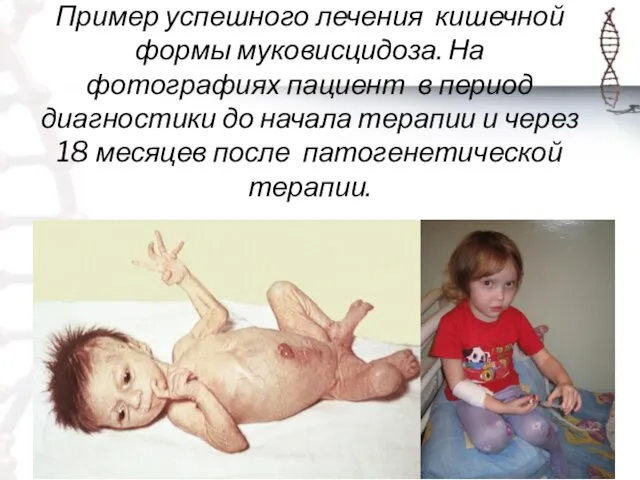
Пример успешного лечения кишечной формы муковисцидоза. На фотографиях пациент в период
Пример успешного лечения кишечной формы муковисцидоза. На фотографиях пациент в период
 Привлечение клиентов с выставок. Выстроить бизнес-процесс по привлечению клиентов с выставок
Привлечение клиентов с выставок. Выстроить бизнес-процесс по привлечению клиентов с выставок A little bit about Spimun
A little bit about Spimun Momente
Momente Имануил Валлерстайн и Теда Скопкол. Историческая ориентация в марксизме
Имануил Валлерстайн и Теда Скопкол. Историческая ориентация в марксизме Эколого-благотворительный проект #Добрые крышечки
Эколого-благотворительный проект #Добрые крышечки 1_Предмет_і_зміст_ТМФВ_Наукові_основи_2 (3)
1_Предмет_і_зміст_ТМФВ_Наукові_основи_2 (3) Презентация День Знаний
Презентация День Знаний Отдел инвестиционной деятельности Управления инвестиционной и инновационной деятельности
Отдел инвестиционной деятельности Управления инвестиционной и инновационной деятельности Строительство тепличных комплексов. Организация тепличного хозяйства в городских округах
Строительство тепличных комплексов. Организация тепличного хозяйства в городских округах Устройства СЦБ и связи при движении поездов
Устройства СЦБ и связи при движении поездов Национальная стратегия противодействия корупции
Национальная стратегия противодействия корупции Формула удачи: улыбка - настроение - вера в себя - результат. Урок русского языка
Формула удачи: улыбка - настроение - вера в себя - результат. Урок русского языка Электромеханикалы көмірлі бұйымдар мен материалдар
Электромеханикалы көмірлі бұйымдар мен материалдар Урок Краеведения 8 класс
Урок Краеведения 8 класс Основные принципы построения систем ввода вывода и интерфейсов
Основные принципы построения систем ввода вывода и интерфейсов презентация волшебный мир театра
презентация волшебный мир театра Портфолио учителя математики Водяковой В. В.
Портфолио учителя математики Водяковой В. В. Привлечение финансирования для создания объектов инфраструктуры
Привлечение финансирования для создания объектов инфраструктуры Прессование изделий из керамических порошков
Прессование изделий из керамических порошков Новые религиозные движения
Новые религиозные движения Качество процесса инсталляционных и установочных работ ДКС МРФ Урал ПАО Ростелеком
Качество процесса инсталляционных и установочных работ ДКС МРФ Урал ПАО Ростелеком Годовой отчет о деятельности организации за 2021.pdf [Восстановленный]
Годовой отчет о деятельности организации за 2021.pdf [Восстановленный] Мировые религии
Мировые религии Республика Беларусь. Теория государственной службы
Республика Беларусь. Теория государственной службы Вируальная выставка Книжные новинки
Вируальная выставка Книжные новинки Презентация к уроку географии Население мира 10 класс
Презентация к уроку географии Население мира 10 класс Как научиться управлять классом
Как научиться управлять классом Поезда. Российские Железные Дороги
Поезда. Российские Железные Дороги