- Биоинформатика. Дендрограммы. Молекулярная филогения. (Тема 6)

Содержание
- 2. Дендрограммы Молекулярная филогения
- 3. Графы и деревья Граф — это простая диаграмма (абстрактная структура), применяемая для представления отношений между элементами
- 4. Терминология Узел (node) — точка разделения предковой последовательности (вида, популяции) на две независимо эволюционирующие. Соответствует внутренней
- 5. Зачем нужны деревья? Биологические задачи: сравнение 3-х и более объектов (кто на кого более похож ....
- 6. Филогенетическое дерево (древо) Филогения - раздел биологии, изучающий родственные взаимоотношения разных групп живых организмов. Молекулярная филогения
- 7. OTU HTU (hypothetical taxonomic unit)
- 8. Какие бывают деревья? Бинарное (разрешённое) (в один момент времени может произойти только одно событие ) Небинарное
- 9. Какие бывают деревья? Укорененное дерево (rooted tree) отражает направление эволюции Неукорененное (бескорневое) дерево (unrooted tree) показывает
- 10. Rooting
- 11. 3 OTUs ⇒ 1 неукорененное дерево 3 укорененных деревьев
- 12. D C A B 4 OTUs ⇒ 3 неукорененных филогенетических деревьев D B A C
- 15. 4 OTUs ⇒ 15 укорененных деревьев
- 16. Количество Количество Количество OTU укорененных неукорененных 2 1 1 3 3 1 4 15 3 5
- 17. Рутинная процедура, или как строят деревья? Составление выборки последовательностей Множественное выравнивание Построение дерева фрагмент записи в
- 18. Рутинная процедура, или как строят деревья? Составление выборки последовательностей Множественное выравнивание (или всё-таки попарное) Построение дерева
- 19. Множественное выравнивание Matches
- 20. Multiple Alignment Matches Mismatches
- 21. Multiple Alignment Matches Mismatches Gaps
- 22. Seq 1 A G C G A G Seq 2 G C G G A C
- 23. Distance Matrix* * Units: количество замен нуклеотидов на 1000
- 24. Шаг 4: построение филогенетического дерева
- 25. Как выбирать последовательности для дерева? Кроме случаев очень близких последовательностей, проще работать с белками (а не
- 26. Самое главное – хорошее выравнивание! Максимальный вклад в финальное дерево: нельзя построить хорошее дерево по плохому
- 27. Основные алгоритмы построения филогенетических деревьев Методы, основанные на оценке расстояний (матричные методы): UPGMA (кластеризация) Neighbor-joining Наибольшего
- 28. Пример матрицы расстояний 1 2 3 4 5 6 7 8 0.00 10.53 9.77 12.78 12.03
- 29. Как понимать расстояние между объектами? Как время, в течение которого они эволюционировали Как число «эволюционных событий»
- 31. Гипотеза «молекулярных часов» (E.Zuckerkandl, L.Pauling, 1962) За равное время во всех ветвях эволюции данного гена\белка накапливается
- 32. UPGMA Unweighted Pair Group Method with Arithmetic Mean разновидность кластерного метода Расстояние между кластерами вычисляется как
- 34. Mon-Hum Monkey Human Spinach Mosquito Rice Дистанция между человеком и обезьяной минимальна. Эти группы объединяются в
- 35. Редуцированная матрица дистанций
- 36. Mon-Hum Monkey Human Spinach Mosquito Rice Spi-Ric
- 37. Human Mosquito Mon-Hum Monkey Spinach Rice Mos-Mon-Hum Spi-Ric Mos-Mon-Hum-Spi-Ric
- 39. Недостатки UPGMA Алгоритм строит ультраметрическое дерево – скорость эволюции предполагается одинаковой для всех ветвей дерева. Использовать
- 40. Метод ближайших соседей (Neighbor-joining, NJ) Строит неукоренённое дерево Может работать с большим количеством данных Достаточно быстрый
- 41. Метод Neighbor-joining Рисуем «звездное» дерево и будем «отщипывать» от него по паре листьев Пусть ui =
- 42. Метод ближайших соседей (Neighbor-joining, NJ) 2. Кластер (i, j) – новый узел дерева Расстояние от i
- 43. Maximum Parsimony (MP)
- 44. Input: MSA для n последовательностей, одна последовательность для каждого вида. AAAAATC AAAAAAG CCCCCCG AAAAATC AAAAAAG CCCCCCG
- 45. Как изобразить дерево? Топология дерева Топология дерева — только листья, узлы, (корень) и связывающие их ветви
- 46. Bacterium 1 Bacterium 3 Bacterium 2 Eukaryote 1 Eukaryote 4 Eukaryote 3 Eukaryote 2 Bacterium 1
- 47. Какие on-line программы строят деревья? ClustalW. “Tree type” – nj, phylip: строит только методом NJ, но
- 48. MEGA: филогенетический анализ последовательностей http://www.megasoftware.net/
- 49. Эволюция – исторический процесс. Из 8,200,794,532,637,891,559,375 деревьев для 20 OTUs, 1 является верным и 8,200,794,532,637,891,559,374 неверны.
- 51. Скачать презентацию

Дендрограммы
Молекулярная филогения
Дендрограммы
Молекулярная филогения

Графы и деревья
Граф — это простая диаграмма (абстрактная структура), применяемая для
Графы и деревья
Граф — это простая диаграмма (абстрактная структура), применяемая для
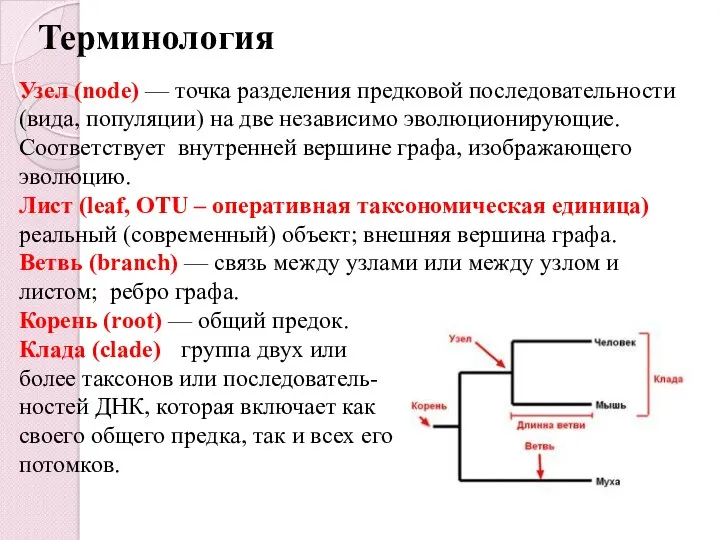
Терминология
Узел (node) — точка разделения предковой последовательности
(вида, популяции) на две независимо
Терминология
Узел (node) — точка разделения предковой последовательности (вида, популяции) на две независимо
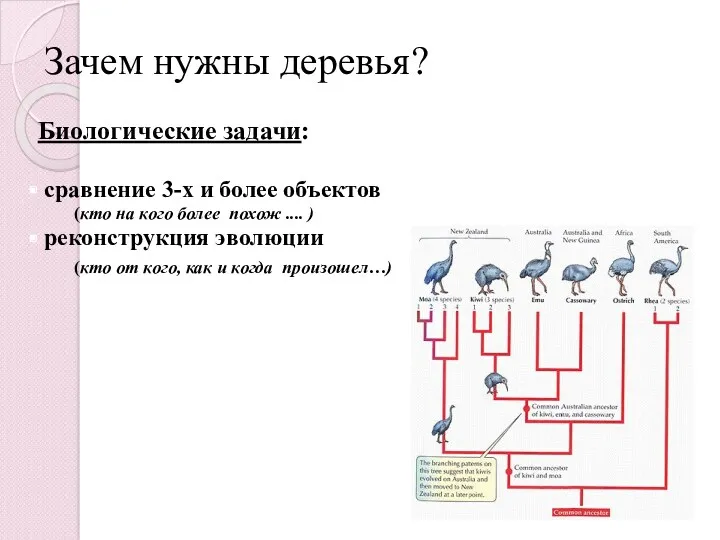
Зачем нужны деревья?
Биологические задачи:
сравнение 3-х и более объектов
(кто на
Зачем нужны деревья?
Биологические задачи:
сравнение 3-х и более объектов
(кто на

Филогенетическое дерево (древо)
Филогения - раздел биологии, изучающий родственные взаимоотношения разных групп
Филогенетическое дерево (древо)
Филогения - раздел биологии, изучающий родственные взаимоотношения разных групп
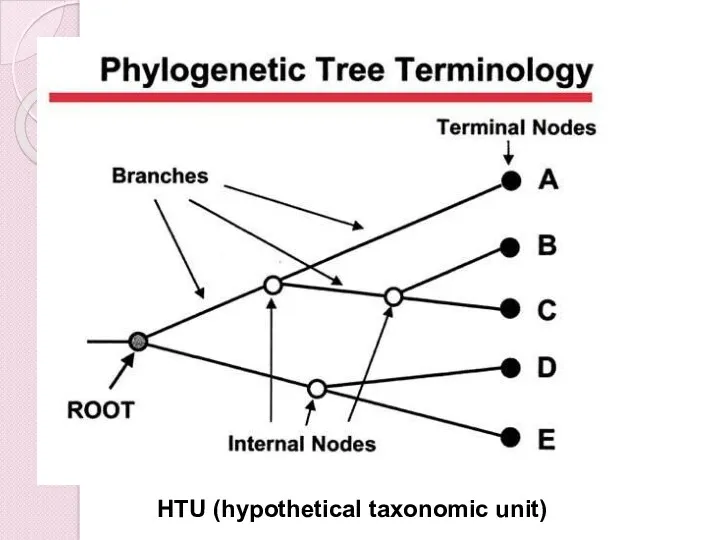
OTU
HTU (hypothetical taxonomic unit)
OTU
HTU (hypothetical taxonomic unit)
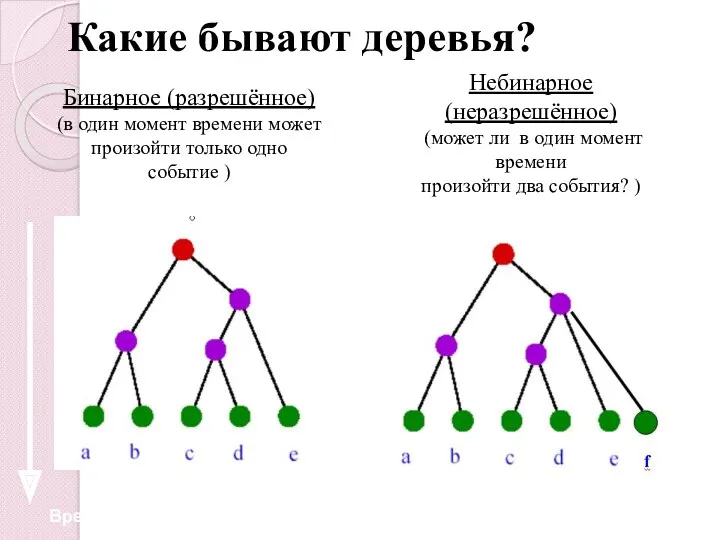
Какие бывают деревья?
Бинарное (разрешённое)
(в один момент времени может
произойти только одно
Какие бывают деревья?
Бинарное (разрешённое)
(в один момент времени может
произойти только одно
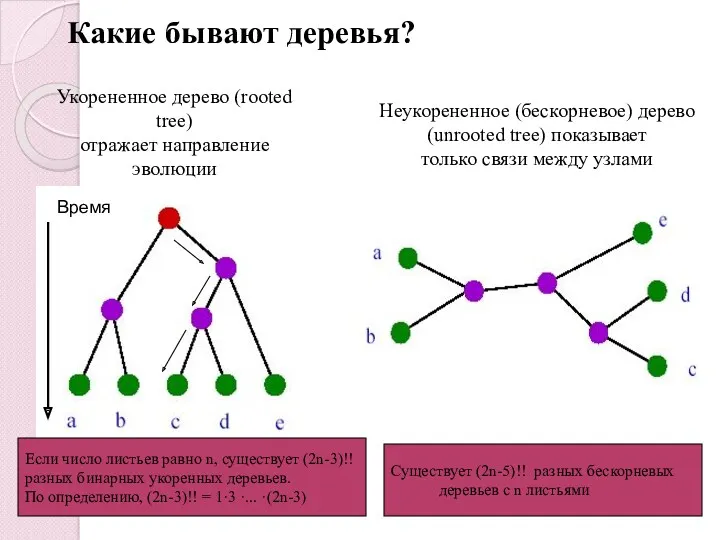
Какие бывают деревья?
Укорененное дерево (rooted tree)
отражает направление эволюции
Неукорененное (бескорневое) дерево
(unrooted tree)
Какие бывают деревья?
Укорененное дерево (rooted tree)
отражает направление эволюции
Неукорененное (бескорневое) дерево (unrooted tree)
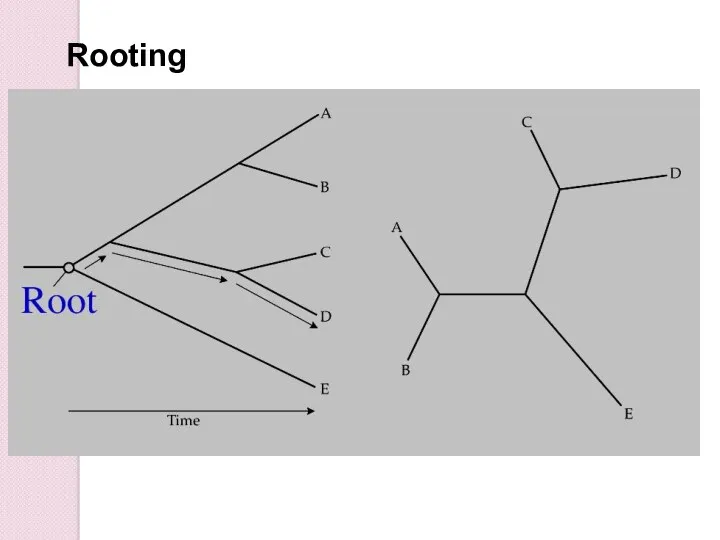
Rooting
Rooting
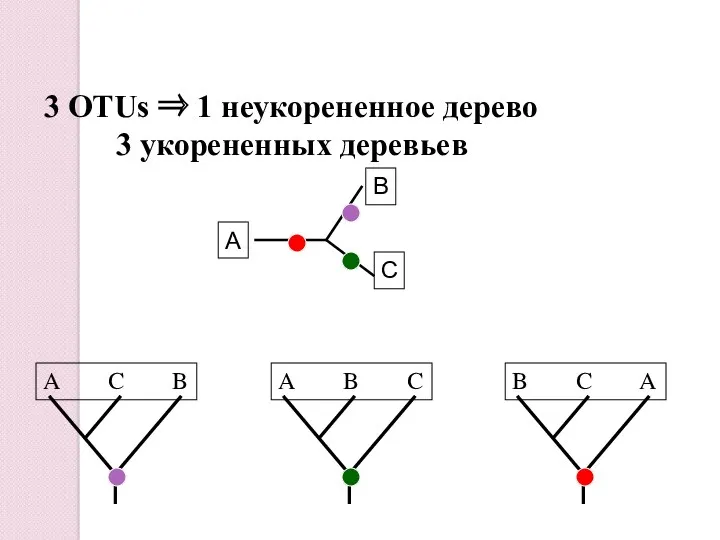
3 OTUs ⇒ 1 неукорененное дерево
3 укорененных деревьев
3 OTUs ⇒ 1 неукорененное дерево
3 укорененных деревьев
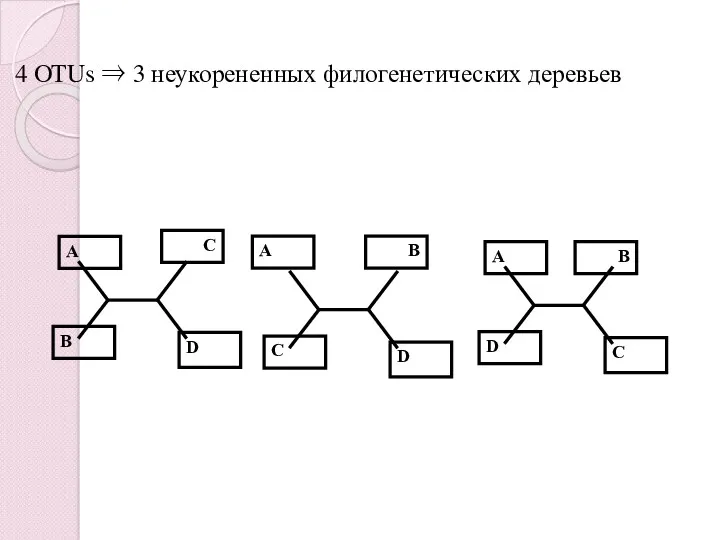
D
C
A
B
4 OTUs ⇒ 3 неукорененных филогенетических деревьев
D
B
A
C
D
C
A
B
4 OTUs ⇒ 3 неукорененных филогенетических деревьев
D
B
A
C

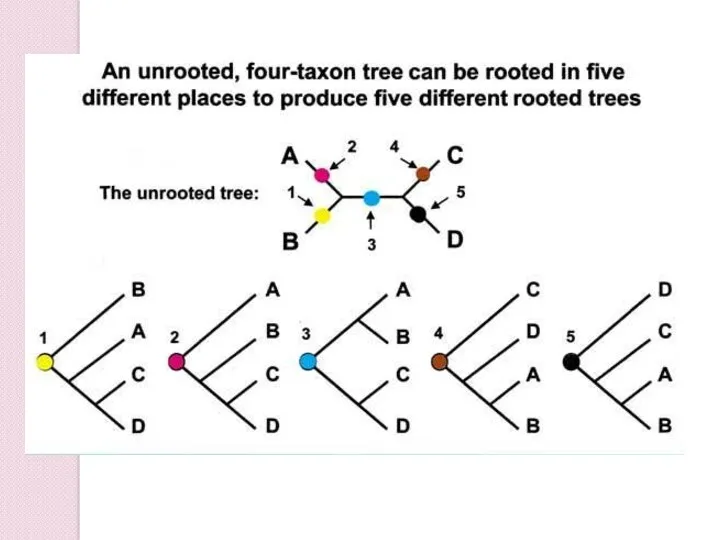
4 OTUs ⇒
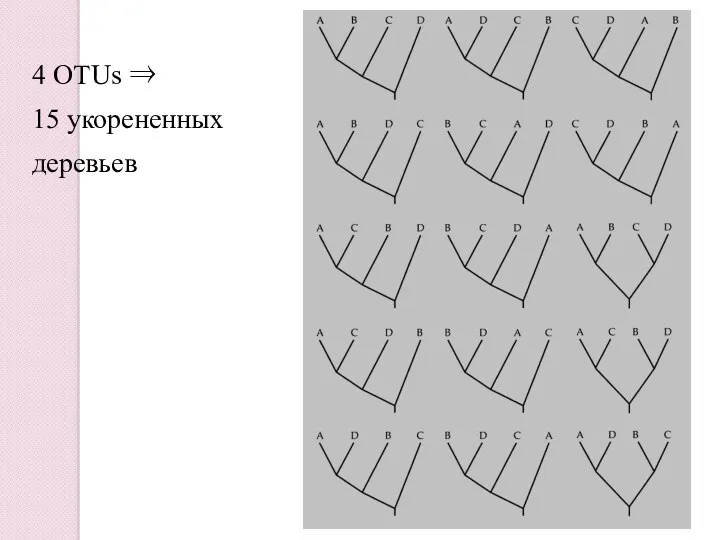
15 укорененных деревьев
4 OTUs ⇒
15 укорененных деревьев

Количество Количество Количество
OTU укорененных неукорененных
2 1 1
3 3 1
4 15 3
5 105 15
6 954 105
7 10,395 954
8 135,135 10,395
9 2,027,025 135,135
10
Количество Количество Количество
OTU укорененных неукорененных
2 1 1
3 3 1
4 15 3
5 105 15
6 954 105
7 10,395 954
8 135,135 10,395
9 2,027,025 135,135
10
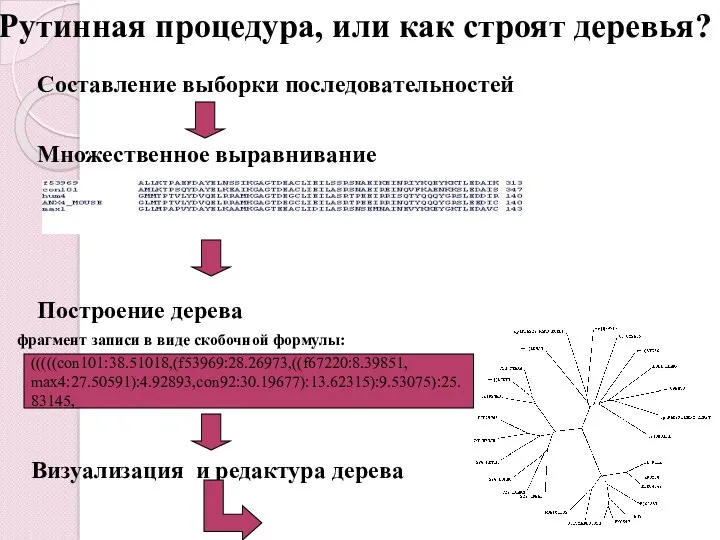
Рутинная процедура, или как строят деревья?
Составление выборки последовательностей
Множественное выравнивание
Рутинная процедура, или как строят деревья?
Составление выборки последовательностей
Множественное выравнивание
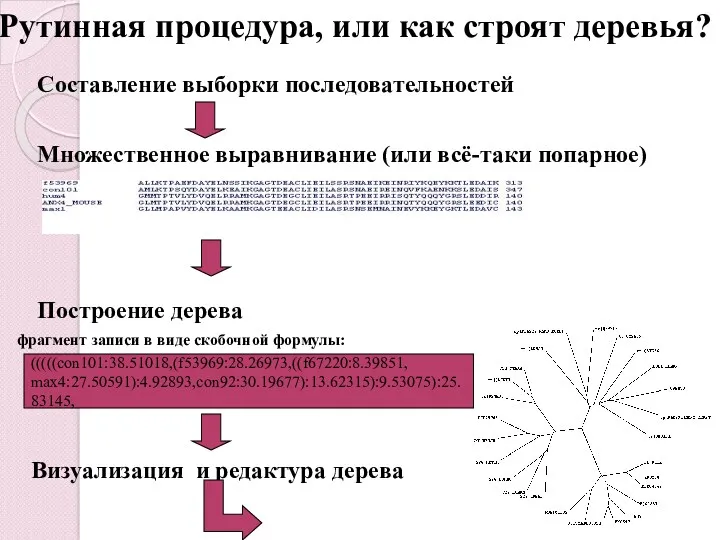
Рутинная процедура, или как строят деревья?
Составление выборки последовательностей
Множественное выравнивание
Рутинная процедура, или как строят деревья?
Составление выборки последовательностей
Множественное выравнивание
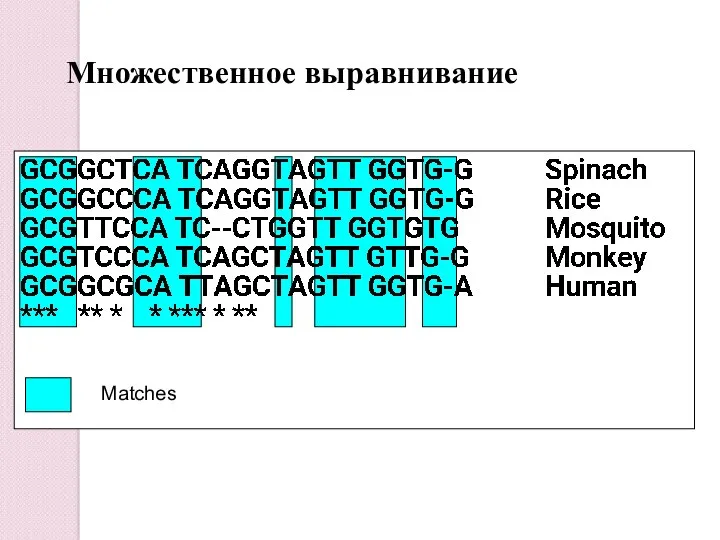
Множественное выравнивание
Matches
Множественное выравнивание
Matches
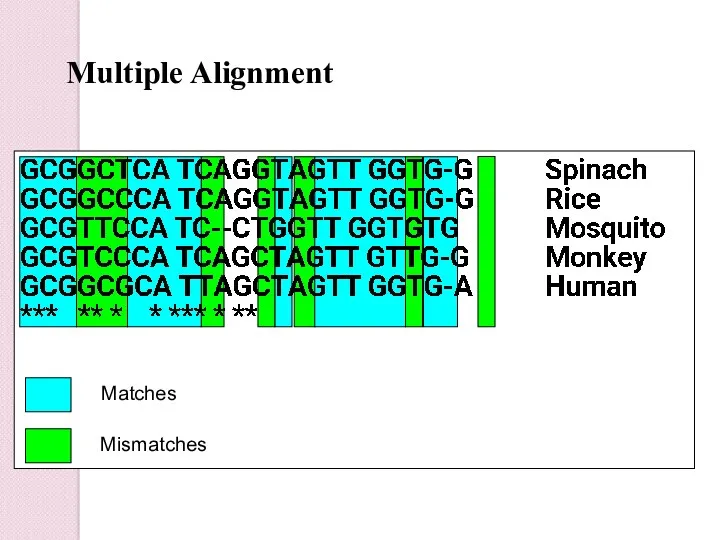
Multiple Alignment
Matches
Mismatches
Multiple Alignment
Matches
Mismatches
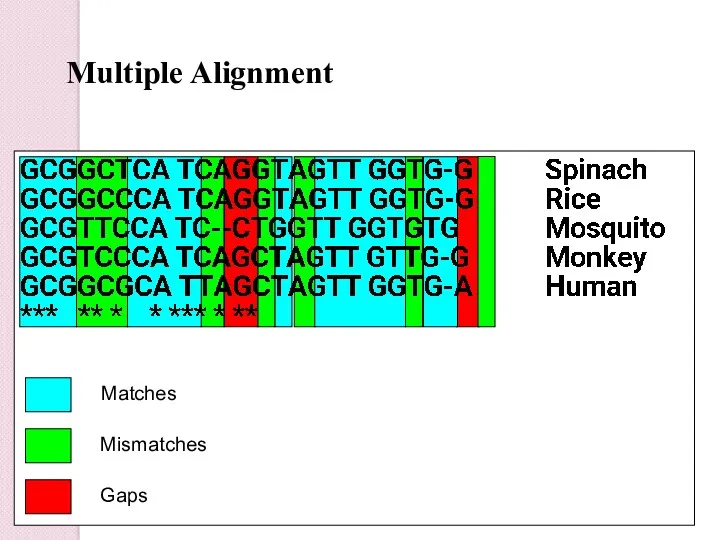
Multiple Alignment
Matches
Mismatches
Gaps
Multiple Alignment
Matches
Mismatches
Gaps
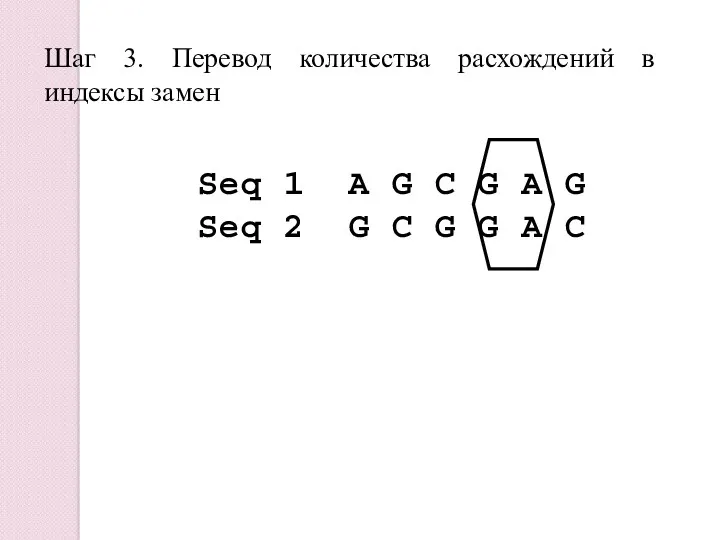
Seq 1 A G C G A G
Seq 2 G C
Seq 1 A G C G A G
Seq 2 G C
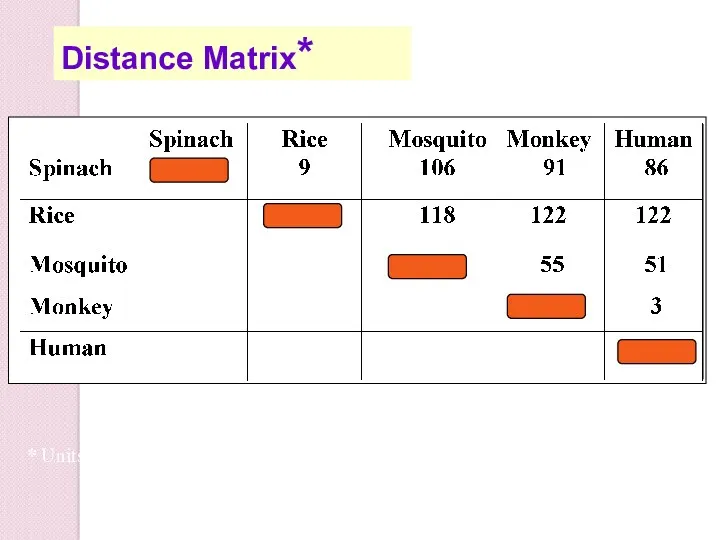
Distance Matrix*
* Units: количество замен нуклеотидов на 1000
Distance Matrix*
* Units: количество замен нуклеотидов на 1000
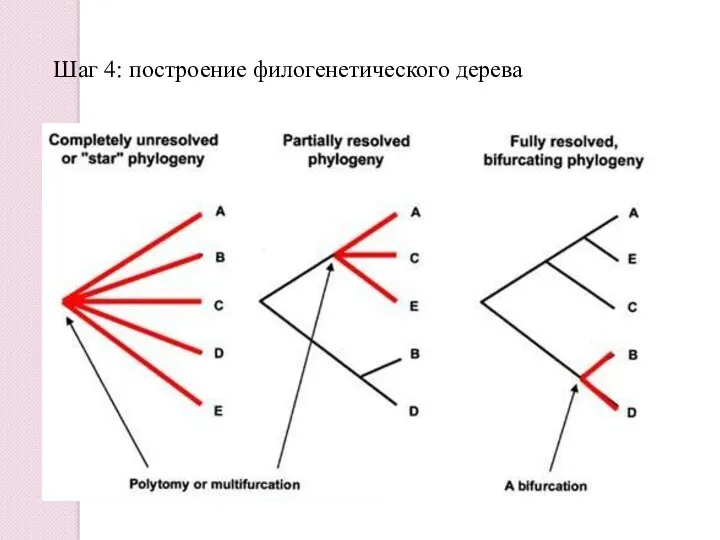
Шаг 4: построение филогенетического дерева
Шаг 4: построение филогенетического дерева

Как выбирать последовательности для дерева?
Кроме случаев очень близких последовательностей, проще работать
Как выбирать последовательности для дерева?
Кроме случаев очень близких последовательностей, проще работать

Самое главное – хорошее выравнивание!
Максимальный вклад в финальное дерево: нельзя построить
Самое главное – хорошее выравнивание!
Максимальный вклад в финальное дерево: нельзя построить
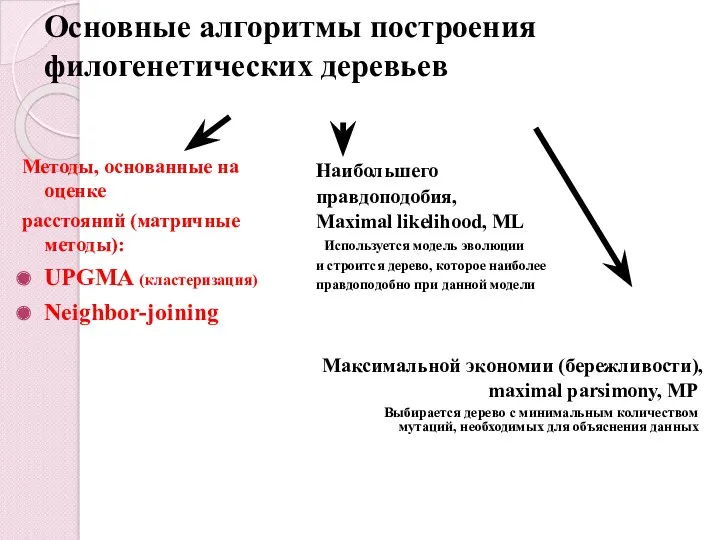
Основные алгоритмы построения филогенетических деревьев
Методы, основанные на оценке
расстояний (матричные методы):
UPGMA
Основные алгоритмы построения филогенетических деревьев
Методы, основанные на оценке
расстояний (матричные методы):
UPGMA

Пример матрицы расстояний
1 2 3 4 5 6 7 8
Пример матрицы расстояний
1 2 3 4 5 6 7 8

Как понимать расстояние между объектами?
Как время, в течение которого они
Как понимать расстояние между объектами?
Как время, в течение которого они

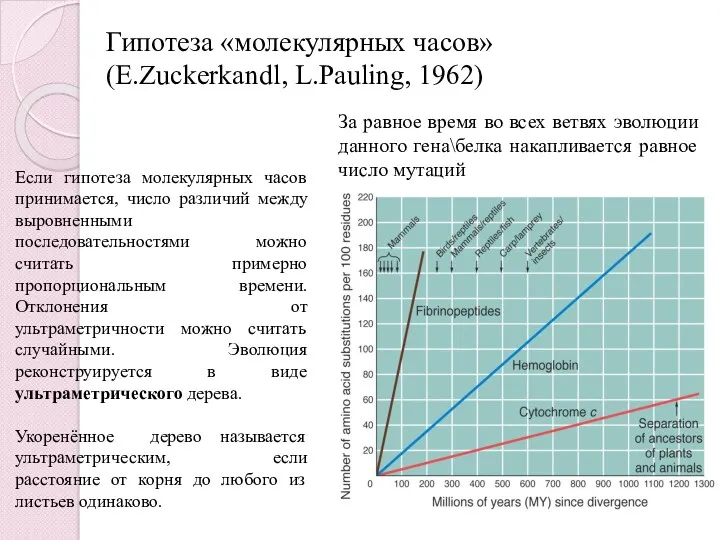
Гипотеза «молекулярных часов»
(E.Zuckerkandl, L.Pauling, 1962)
За равное время во всех ветвях эволюции
Гипотеза «молекулярных часов»
(E.Zuckerkandl, L.Pauling, 1962)
За равное время во всех ветвях эволюции

UPGMA
Unweighted Pair Group Method with Arithmetic Mean
разновидность кластерного метода
Расстояние между кластерами
UPGMA
Unweighted Pair Group Method with Arithmetic Mean
разновидность кластерного метода
Расстояние между кластерами
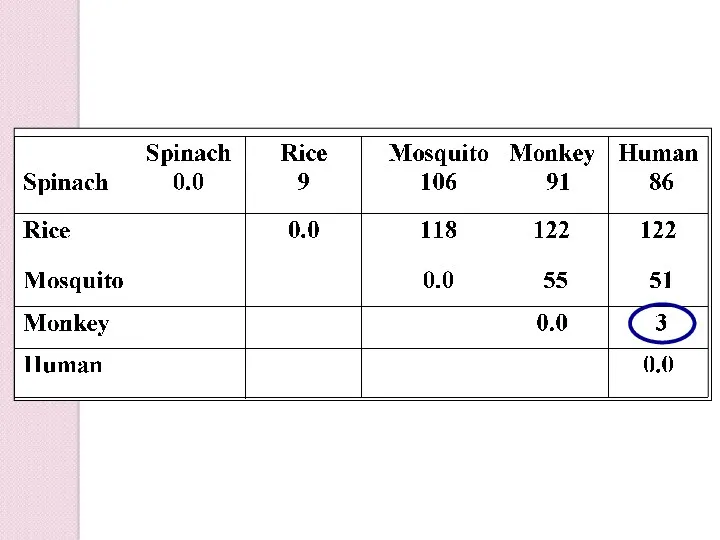

Mon-Hum
Monkey
Human
Spinach
Mosquito
Rice
Дистанция между человеком и обезьяной минимальна. Эти группы объединяются в Monkey-Human,
Mon-Hum
Monkey
Human
Spinach
Mosquito
Rice
Дистанция между человеком и обезьяной минимальна. Эти группы объединяются в Monkey-Human,

Редуцированная матрица дистанций
Редуцированная матрица дистанций
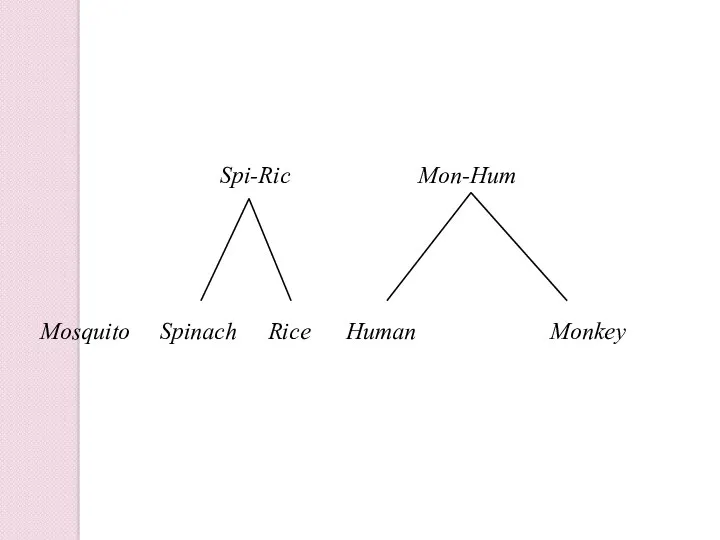
Mon-Hum
Monkey
Human
Spinach
Mosquito
Rice
Spi-Ric
Mon-Hum
Monkey
Human
Spinach
Mosquito
Rice
Spi-Ric
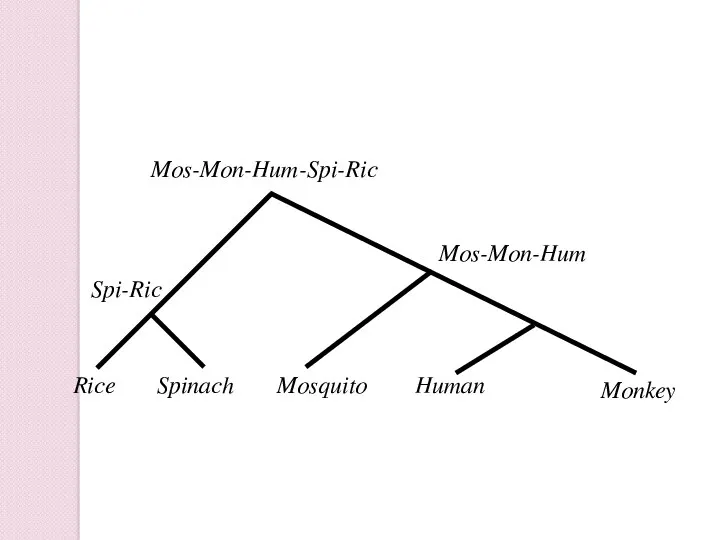
Human
Mosquito
Mon-Hum
Monkey
Spinach
Rice
Mos-Mon-Hum
Spi-Ric
Mos-Mon-Hum-Spi-Ric
Human
Mosquito
Mon-Hum
Monkey
Spinach
Rice
Mos-Mon-Hum
Spi-Ric
Mos-Mon-Hum-Spi-Ric
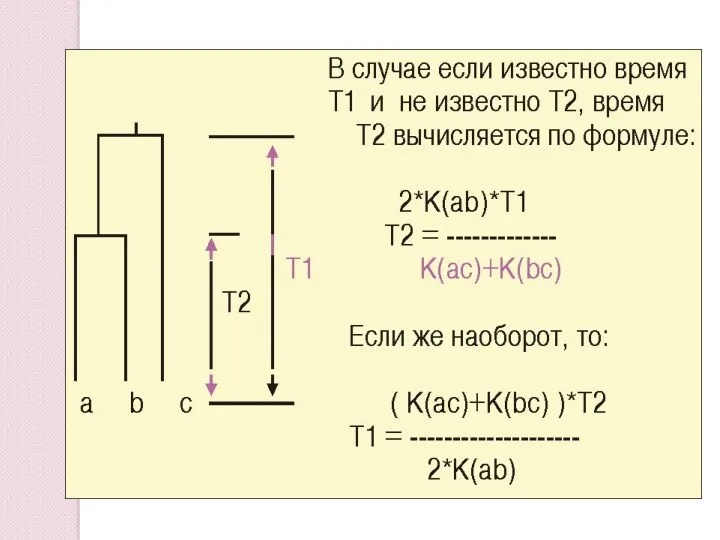
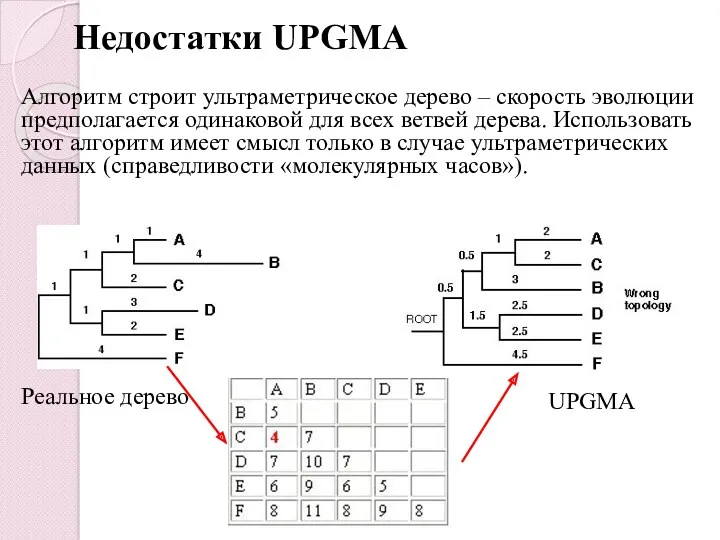
Недостатки UPGMA
Алгоритм строит ультраметрическое дерево – скорость эволюции
предполагается одинаковой
Недостатки UPGMA
Алгоритм строит ультраметрическое дерево – скорость эволюции предполагается одинаковой

Метод ближайших соседей
(Neighbor-joining, NJ)
Строит неукоренённое дерево
Может работать с большим количеством
Метод ближайших соседей
(Neighbor-joining, NJ)
Строит неукоренённое дерево
Может работать с большим количеством
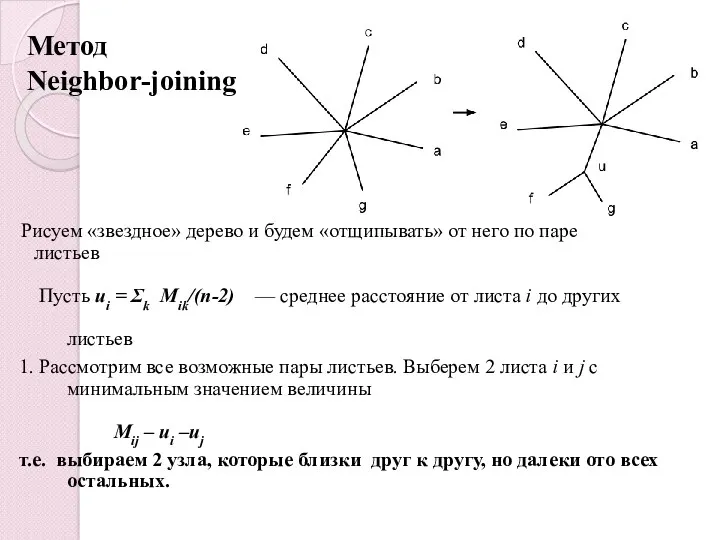
Метод
Neighbor-joining
Рисуем «звездное» дерево и будем «отщипывать» от него по
Метод
Neighbor-joining
Рисуем «звездное» дерево и будем «отщипывать» от него по
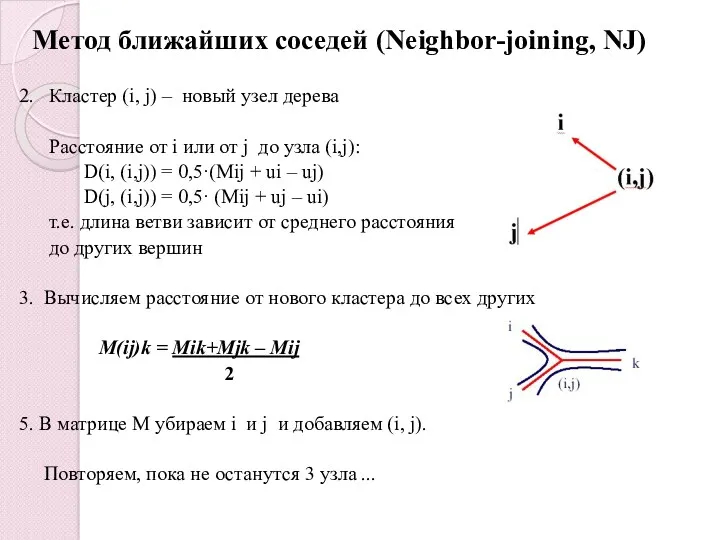
Метод ближайших соседей (Neighbor-joining, NJ)
2. Кластер (i, j) – новый узел
Метод ближайших соседей (Neighbor-joining, NJ)
2. Кластер (i, j) – новый узел
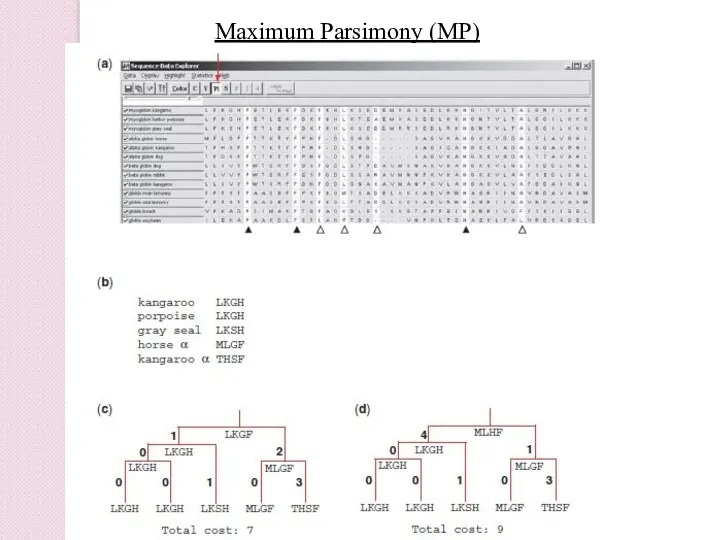
Maximum Parsimony (MP)
Maximum Parsimony (MP)
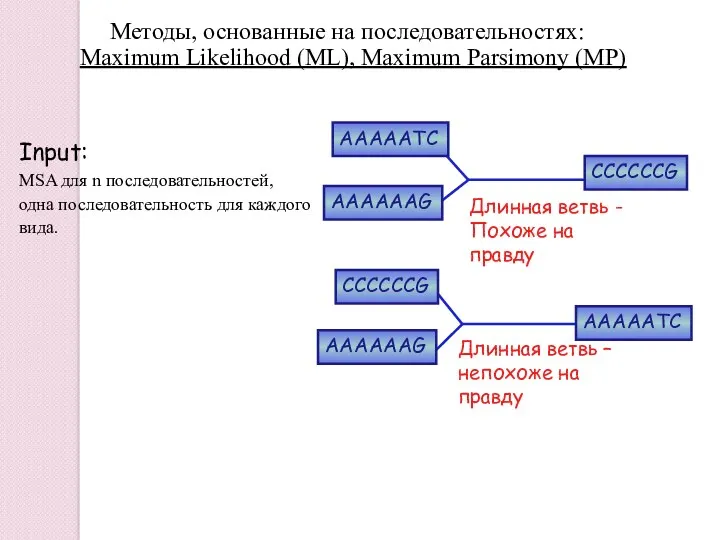
Input:
MSA для n последовательностей, одна последовательность для каждого вида.
AAAAATC
AAAAAAG
CCCCCCG
AAAAATC
AAAAAAG
CCCCCCG
Длинная ветвь
Input:
MSA для n последовательностей, одна последовательность для каждого вида.
AAAAATC
AAAAAAG
CCCCCCG
AAAAATC
AAAAAAG
CCCCCCG
Длинная ветвь
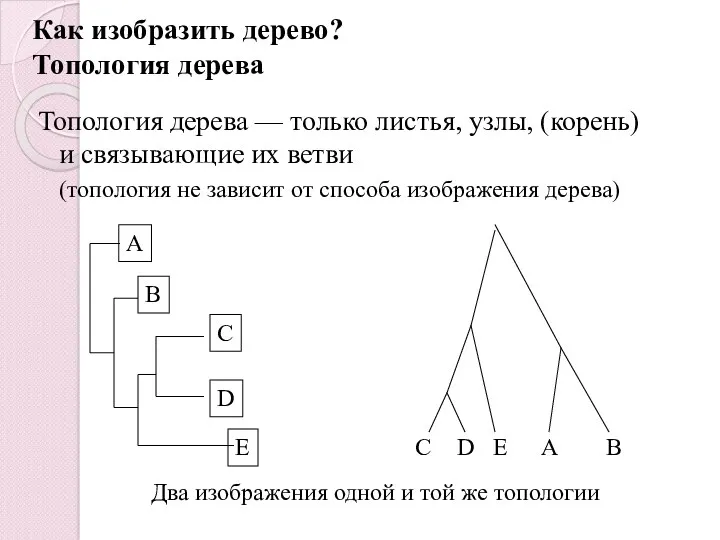
Как изобразить дерево?
Топология дерева
Топология дерева — только листья, узлы, (корень)
Как изобразить дерево?
Топология дерева
Топология дерева — только листья, узлы, (корень)
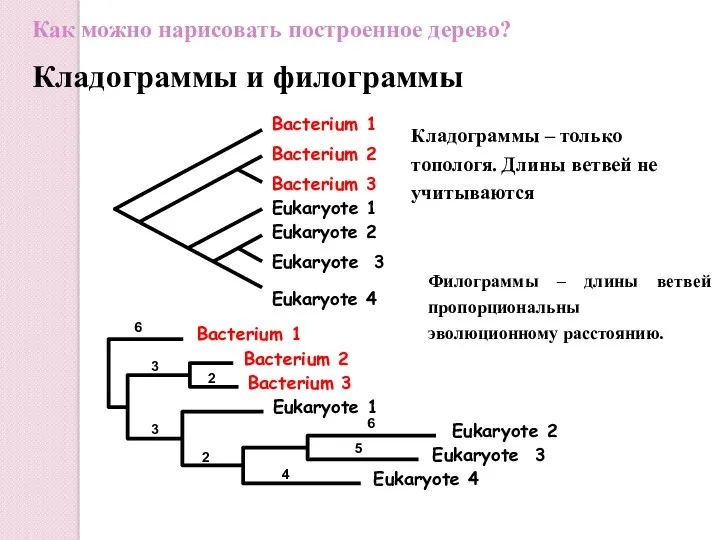
Bacterium 1
Bacterium 3
Bacterium 2
Eukaryote 1
Eukaryote 4
Eukaryote 3
Eukaryote 2
Bacterium 1
Bacterium 3
Bacterium 2
Eukaryote
Bacterium 1
Bacterium 3
Bacterium 2
Eukaryote 1
Eukaryote 4
Eukaryote 3
Eukaryote 2
Bacterium 1
Bacterium 3
Bacterium 2
Eukaryote

Какие on-line программы строят деревья?
ClustalW. “Tree type” – nj, phylip: строит
Какие on-line программы строят деревья?
ClustalW. “Tree type” – nj, phylip: строит

MEGA: филогенетический анализ последовательностей
http://www.megasoftware.net/
MEGA: филогенетический анализ последовательностей
http://www.megasoftware.net/

Эволюция – исторический процесс.
Из 8,200,794,532,637,891,559,375 деревьев для 20 OTUs, 1 является
Эволюция – исторический процесс.
Из 8,200,794,532,637,891,559,375 деревьев для 20 OTUs, 1 является
 Антропогенез. Мартышкообразные: секрет успеха. (часть 8)
Антропогенез. Мартышкообразные: секрет успеха. (часть 8) Лист. Его строение и значение
Лист. Его строение и значение Растения луга Вологодской области, занесённые в красную книгу
Растения луга Вологодской области, занесённые в красную книгу Секреты природы. Лес - наше богатство. Занятие №1
Секреты природы. Лес - наше богатство. Занятие №1 Молекулярно-генетические механизмы развития корня
Молекулярно-генетические механизмы развития корня Женская половая система
Женская половая система Происхождение и начальные этапы развития жизни на Земле
Происхождение и начальные этапы развития жизни на Земле Отруйні гриби
Отруйні гриби Общие представления о биомолекулах. Химическая эволюция. Прокариоты и эукариоты. Структура клеток. Вирусы
Общие представления о биомолекулах. Химическая эволюция. Прокариоты и эукариоты. Структура клеток. Вирусы Презентация Отряд Блохи
Презентация Отряд Блохи Квітка. Будова квітки
Квітка. Будова квітки Круговорот углерода в природе
Круговорот углерода в природе Подводный мир
Подводный мир Тип Плоские Черви
Тип Плоские Черви Строение клетки
Строение клетки Как зимуют птицы
Как зимуют птицы Animals and Their Habitat
Animals and Their Habitat Вирусы – неклеточные формы жизни
Вирусы – неклеточные формы жизни Түйелер шөлге де, қатты аязға да төзімді, күшті көлік малы
Түйелер шөлге де, қатты аязға да төзімді, күшті көлік малы Покриви тіла тварин
Покриви тіла тварин Органы дыхания беспозвоночных и позвоночных животных
Органы дыхания беспозвоночных и позвоночных животных Пищеварительная система
Пищеварительная система Человек. Глава 1
Человек. Глава 1 Магнитные поля. История магнитобиологии
Магнитные поля. История магнитобиологии Видообразование. Результаты микроэволюции
Видообразование. Результаты микроэволюции Животный и растительный мир в пустынях
Животный и растительный мир в пустынях СПИД - сведи вероятность к нулю(презентация)
СПИД - сведи вероятность к нулю(презентация) Виды тканей растений и животных
Виды тканей растений и животных