Мутации. Классификации. Мутации и мобильные элементы. Мобильные элементы генома, классификация и их генетическая роль презентация
- Мутации. Классификации. Мутации и мобильные элементы. Мобильные элементы генома, классификация и их генетическая роль

Содержание
- 2. Мутации Мутационная теория— раздел генетики, закладывающий основы генетической изменчивости и эволюции. Мутации – это качественные изменения
- 3. Классификация мутаций По характеру изменения генома (по уровню организации генетического материала, затронутого изменениями): Геномные – изменение
- 4. Классификация мутаций по их молекулярной природе 1. Точковые мутации (замена нуклеотидов): (с химической точки зрения) транзиции:
- 5. Точковые мутации в кодирующей области синонимичные несинонимичные: миссенс-мутации – смена АМК: консервативные - заменам АМК в
- 6. Серповидно-клеточная анемия Причина: Генные мутации полипептидной цепочки гемоглобина; в результате наблюдается преждевременный гемолиз и распад эритроцитов,
- 7. Талассемия Причина: Мутация регуляторных глобиновых генов, вследствие чего наблюдается нестабильный синтез и дисфункция матричной РНК. Дефицит
- 8. nonsense-mediated mRNA decay Нонсенс-мутация (появление стоп кодона) ? Преждевременная терминация трансляции ? Синтез укороченного и скорее
- 9. затрагивающие регуляторную область незначащие (нейтральные) _____________________________ исчезновение или появление сайта сплайсинга (альтернативный сплайсинг свойственен 80% генов
- 10. Классификация мутаций по их молекулярной природе 2. Мутации сдвига рамки считывания Indels – термин для обозначения
- 11. Амавротическая идиотия (болезнь Тея-Сакса) Причина: Болезнь связана с нарушением липидного обмена. Для нее характерно отложение в
- 12. Классификация мутаций по их молекулярной природе 3. Делеция и инсерция промежутков Транспозиция – перемещение участка ДНК
- 13. Муковисцидоз Причина: Поражение экзокринных желез и железистых клеток организма: секретирующих клеток бронхов, поджелудочной железы, кишечника, потовых
- 14. Классификация мутаций по их молекулярной природе 4. Хромосомные мутации. Хромосомные перестройки делеции (утрата участка хромосомы) инверсии
- 15. Классификация мутаций по их молекулярной природе 4. Хромосомные мутации. Изменениях плоидности, то есть количества хромосом в
- 16. Классификация мутаций по месту возникновения: Ядерные Цитоплазматические (мутация неядерных генов – митохондрий и хлоропластов) __________________________________________________ В
- 17. Классификация мутаций по характеру мутагенного фактора: Спонтанные ошибки ДНК-полимеразы при репликации ДНК нарушение в мейозе неправильный
- 18. Биологические и физические мутагены Биологические мутагены специфические последовательности ДНК - транспозоны; некоторые вирусы (вирус кори, краснухи,
- 19. Химические мутагены некоторые алкалоиды: колхицин — один из самых распространённых в селекции мутагенов, винкамин, подофиллотоксин; окислители
- 20. Классификация мутаций по функции молекулярного продукта мутантных аллелей (Герман Меллер): аморфные мутации – молекулярная функция отсутствуют
- 21. Классификация мутаций по действию на фенотип: летальные мутации – носитель нежизнеспособен, фенотипа нет. морфологические мутации –
- 22. Классификация мутаций по действию на приспособленность (жизнеспособность, плодовитость и скорость индивидуального развития) летальные вредные полезные нейтральные
- 23. Наследственные болезни человека Наследственные болезни, которые развиваются только при наличии мутантного гена; они передаются из поколения
- 24. Наследственные болезни человека Все наследственные болезни делятся на три группы: Генные (моногенные - в основе патологии
- 25. Гемофилия Причина: Наследственный дефицит плазменного фактора свертывания крови в связи с прямой мутацией гена, локализованного в
- 26. Синдром Марфана (арахнодактилия) Причина: Изменение обмена мукополисахаридов приводит к нарушению образования коллагена. Тип наследования: Аутосомно-доминантный. Клиника:
- 27. Нейрофиброматоз (болезнь Реклингхаузена) Причина: У больных в печени, почках и слизистой кишечника накапливается большое количество гликогена.
- 28. Синдром Вильямса, «лицо Эльфа» Причина: У детей повышенный уровень кальция в сыворотке крови. Тип наследования: Аутосомно-доминантный.
- 29. Тяжёлый комбинированный иммунодефицит Дэвид Веттер это генетическое заболевание, при котором в результате дефекта одного из генов
- 30. Митохондриальные заболевания Митохондриальный сахарный диабет, сопровождающийся - это сочетание, проявляющееся в раннем возрасте, может быть вызвано
- 31. Болезни, обусловленные нарушением числа аутосом (неполовых) хромосом Синдром Дауна — трисомия по 21 хромосоме, к признакам
- 32. Болезни, связанные с нарушением числа половых хромосом Синдром Шерешевского — Тёрнера — отсутствие одной Х-хромосомы у
- 33. Болезни с наследственной предрасположенностью Выделяют следующие основные группы болезней с наследственной предрасположенностью: врожденные пороки развития (анэнцефалия,
- 34. Мобильные генетические элементы Мобильными генетическими элементами, менее строго называемыми также транспозонами, называются отрезки ДНК генома, способные:
- 35. Класс 1. Ретротранспозоны. Размножаются в геноме посредством копирования и инсерции копий. Копирование идет через РНК-посредник и
- 36. Подкласс LTR-ретротранспозоны LTR – Long Treminal Repeat - длинные (100–5000 пар оснований) прямые терминальные повторы. Они
- 37. Подкласс Non-LTR Retrotransposones LINE (Long Interspersed Elements) –длинные диспергированные (несколько тысяч пар оснований) последовательности ДНК в
- 38. Класс 2. ДНК-транспозоны. ДНК-транспозоны перемещаются по геному посредством транспозиции, эксцизируясь в одном месте и интегрируясь в
- 39. Подкласс IS-элементы IS - insertion sequences - инсерционные последовательности, в своем составе имеют только фермент для
- 40. Класс 3. Пассивные элементы типа fold-back К этому классу относят семейство элементов FB (fold back). Размеры
- 41. Открытие транспозонов у кукурузы В 40-х годах Барбара Мак-Клинток, американский генетик, обнаружила мозаичность окраски зерен у
- 42. У млекопитающих около 44-48% генома составляют транспозоны или их остатки. У человека 44% генома - транспозоны,
- 43. Изменение активности гена: Внедрение мобильных элементов внутрь гена приводит к выключению гена. Может нарушаться регуляция гена,
- 45. Скачать презентацию

Мутации
Мутационная теория— раздел генетики, закладывающий основы генетической изменчивости и эволюции.
Мутации –
Мутации
Мутационная теория— раздел генетики, закладывающий основы генетической изменчивости и эволюции.
Мутации –

Классификация мутаций
По характеру изменения генома (по уровню организации генетического материала,
Классификация мутаций
По характеру изменения генома (по уровню организации генетического материала,
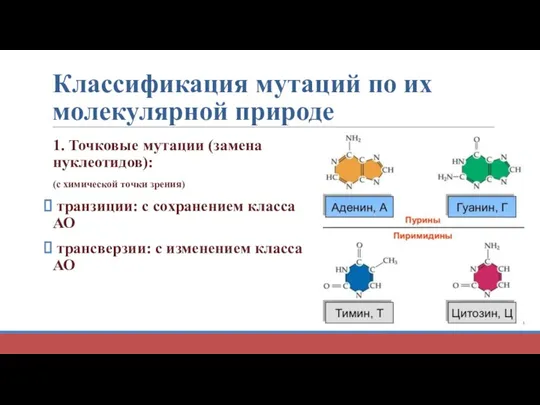
Классификация мутаций по их молекулярной природе
1. Точковые мутации (замена нуклеотидов):
(с химической
Классификация мутаций по их молекулярной природе
1. Точковые мутации (замена нуклеотидов):
(с химической

Точковые мутации в кодирующей области
синонимичные
несинонимичные:
миссенс-мутации – смена
Точковые мутации в кодирующей области
синонимичные
несинонимичные:
миссенс-мутации – смена
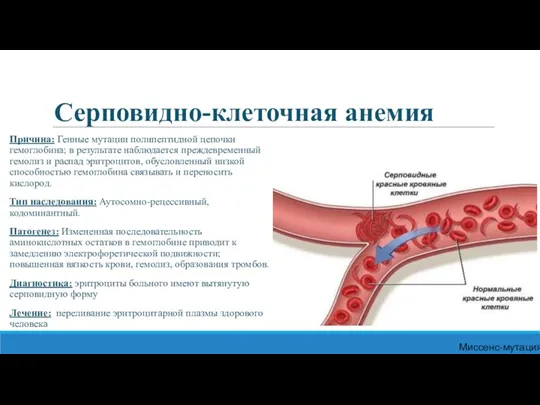
Серповидно-клеточная анемия
Причина: Генные мутации полипептидной цепочки гемоглобина; в результате наблюдается преждевременный
Серповидно-клеточная анемия
Причина: Генные мутации полипептидной цепочки гемоглобина; в результате наблюдается преждевременный

Талассемия
Причина: Мутация регуляторных глобиновых генов, вследствие чего наблюдается нестабильный синтез и
Талассемия
Причина: Мутация регуляторных глобиновых генов, вследствие чего наблюдается нестабильный синтез и

nonsense-mediated mRNA decay
Нонсенс-мутация (появление стоп кодона)
? Преждевременная терминация трансляции
nonsense-mediated mRNA decay
Нонсенс-мутация (появление стоп кодона)
? Преждевременная терминация трансляции

затрагивающие регуляторную область
незначащие (нейтральные)
_____________________________
исчезновение или появление сайта
затрагивающие регуляторную область
незначащие (нейтральные)
_____________________________
исчезновение или появление сайта

Классификация мутаций по их молекулярной природе
2. Мутации сдвига рамки считывания
Indels –
Классификация мутаций по их молекулярной природе
2. Мутации сдвига рамки считывания
Indels –
Амавротическая идиотия
(болезнь Тея-Сакса)
Причина: Болезнь связана с нарушением липидного обмена. Для
Амавротическая идиотия
(болезнь Тея-Сакса)
Причина: Болезнь связана с нарушением липидного обмена. Для

Классификация мутаций по их молекулярной природе
3. Делеция и инсерция промежутков
Транспозиция –
Классификация мутаций по их молекулярной природе
3. Делеция и инсерция промежутков
Транспозиция –

Муковисцидоз
Причина: Поражение экзокринных желез и железистых клеток организма: секретирующих клеток бронхов,
Муковисцидоз
Причина: Поражение экзокринных желез и железистых клеток организма: секретирующих клеток бронхов,

Классификация мутаций по их молекулярной природе
4. Хромосомные мутации.
Хромосомные перестройки
Классификация мутаций по их молекулярной природе
4. Хромосомные мутации.
Хромосомные перестройки

Классификация мутаций по их молекулярной природе
4. Хромосомные мутации.
Изменениях плоидности,
Классификация мутаций по их молекулярной природе
4. Хромосомные мутации.
Изменениях плоидности,

Классификация мутаций по месту возникновения:
Ядерные
Цитоплазматические (мутация неядерных генов
Классификация мутаций по месту возникновения:
Ядерные
Цитоплазматические (мутация неядерных генов

Классификация мутаций по характеру мутагенного фактора:
Спонтанные
ошибки ДНК-полимеразы при
Классификация мутаций по характеру мутагенного фактора:
Спонтанные
ошибки ДНК-полимеразы при

Биологические и физические мутагены
Биологические мутагены
специфические последовательности ДНК -
Биологические и физические мутагены
Биологические мутагены
специфические последовательности ДНК -

Химические мутагены
некоторые алкалоиды: колхицин — один из самых распространённых в
Химические мутагены
некоторые алкалоиды: колхицин — один из самых распространённых в

Классификация мутаций по функции молекулярного продукта мутантных аллелей (Герман Меллер):
аморфные
Классификация мутаций по функции молекулярного продукта мутантных аллелей (Герман Меллер):
аморфные

Классификация мутаций по действию на фенотип:
летальные мутации – носитель нежизнеспособен,
Классификация мутаций по действию на фенотип:
летальные мутации – носитель нежизнеспособен,

Классификация мутаций по действию на приспособленность
(жизнеспособность, плодовитость и скорость индивидуального
Классификация мутаций по действию на приспособленность
(жизнеспособность, плодовитость и скорость индивидуального

Наследственные болезни человека
Наследственные болезни, которые развиваются только при наличии мутантного
Наследственные болезни человека
Наследственные болезни, которые развиваются только при наличии мутантного

Наследственные болезни человека
Все наследственные болезни делятся на три группы:
Генные (моногенные
Наследственные болезни человека
Все наследственные болезни делятся на три группы:
Генные (моногенные

Гемофилия
Причина: Наследственный дефицит плазменного фактора свертывания крови в связи с прямой
Гемофилия
Причина: Наследственный дефицит плазменного фактора свертывания крови в связи с прямой

Синдром Марфана (арахнодактилия)
Причина: Изменение обмена мукополисахаридов приводит к нарушению образования коллагена.
Тип
Синдром Марфана (арахнодактилия)
Причина: Изменение обмена мукополисахаридов приводит к нарушению образования коллагена.
Тип

Нейрофиброматоз (болезнь Реклингхаузена)
Причина: У больных в печени, почках и слизистой кишечника
Нейрофиброматоз (болезнь Реклингхаузена)
Причина: У больных в печени, почках и слизистой кишечника

Синдром Вильямса, «лицо Эльфа»
Причина: У детей повышенный уровень кальция в сыворотке
Синдром Вильямса, «лицо Эльфа»
Причина: У детей повышенный уровень кальция в сыворотке

Тяжёлый комбинированный иммунодефицит
Дэвид Веттер
это генетическое заболевание, при котором в результате дефекта
Тяжёлый комбинированный иммунодефицит
Дэвид Веттер
это генетическое заболевание, при котором в результате дефекта

Митохондриальные заболевания
Митохондриальный сахарный диабет, сопровождающийся - это сочетание, проявляющееся в
Митохондриальные заболевания
Митохондриальный сахарный диабет, сопровождающийся - это сочетание, проявляющееся в

Болезни, обусловленные нарушением числа аутосом (неполовых) хромосом
Синдром Дауна —
Болезни, обусловленные нарушением числа аутосом (неполовых) хромосом
Синдром Дауна —

Болезни, связанные с нарушением числа половых хромосом
Синдром Шерешевского —
Болезни, связанные с нарушением числа половых хромосом
Синдром Шерешевского —

Болезни с наследственной предрасположенностью
Выделяют следующие основные группы болезней с
Болезни с наследственной предрасположенностью
Выделяют следующие основные группы болезней с

Мобильные генетические элементы
Мобильными генетическими элементами, менее строго называемыми также транспозонами, называются
Мобильные генетические элементы
Мобильными генетическими элементами, менее строго называемыми также транспозонами, называются

Класс 1. Ретротранспозоны.
Размножаются в геноме посредством копирования и инсерции копий.
Копирование
Класс 1. Ретротранспозоны.
Размножаются в геноме посредством копирования и инсерции копий.
Копирование

Подкласс LTR-ретротранспозоны
LTR – Long Treminal Repeat - длинные (100–5000 пар оснований)
Подкласс LTR-ретротранспозоны
LTR – Long Treminal Repeat - длинные (100–5000 пар оснований)

Подкласс Non-LTR Retrotransposones
LINE (Long Interspersed Elements) –длинные диспергированные (несколько тысяч пар
Подкласс Non-LTR Retrotransposones
LINE (Long Interspersed Elements) –длинные диспергированные (несколько тысяч пар

Класс 2. ДНК-транспозоны.
ДНК-транспозоны перемещаются по геному посредством транспозиции, эксцизируясь в
Класс 2. ДНК-транспозоны.
ДНК-транспозоны перемещаются по геному посредством транспозиции, эксцизируясь в

Подкласс IS-элементы
IS - insertion sequences - инсерционные последовательности, в своем составе
Подкласс IS-элементы
IS - insertion sequences - инсерционные последовательности, в своем составе

Класс 3. Пассивные элементы типа fold-back
К этому классу относят семейство элементов
Класс 3. Пассивные элементы типа fold-back
К этому классу относят семейство элементов

Открытие транспозонов у кукурузы
В 40-х годах Барбара Мак-Клинток, американский генетик, обнаружила
Открытие транспозонов у кукурузы
В 40-х годах Барбара Мак-Клинток, американский генетик, обнаружила

У млекопитающих около 44-48% генома составляют транспозоны или их остатки.
У
У млекопитающих около 44-48% генома составляют транспозоны или их остатки.
У

Изменение активности гена:
Внедрение мобильных элементов внутрь гена приводит к
Изменение активности гена:
Внедрение мобильных элементов внутрь гена приводит к
 Тип Молюски. Клас Двостулкові
Тип Молюски. Клас Двостулкові Растительный и животный мир Донецкого края
Растительный и животный мир Донецкого края Фізіологія всмоктування і моторна діяльність травного каналу
Фізіологія всмоктування і моторна діяльність травного каналу Fats and oils
Fats and oils Интересные факты
Интересные факты Сучасні критерії виду
Сучасні критерії виду Как распознать живое и неживое
Как распознать живое и неживое Рослини-хижаки. Товстянка звичайна
Рослини-хижаки. Товстянка звичайна Увеличительные приборы. Устройство ручной и штативной лупы, светового микроскопа. Правила работы с микроскопом
Увеличительные приборы. Устройство ручной и штативной лупы, светового микроскопа. Правила работы с микроскопом Водная среда обитания организмов
Водная среда обитания организмов Паразитизм. Виды паразитизма. Паразитология
Паразитизм. Виды паразитизма. Паразитология Жүйке жүйесі. Жүйке жүйесінің мүшелерінің ПРе- және ПОстнатальды дамуы. Адам онтогенезінде функционалды жүйелер туралы түсінік
Жүйке жүйесі. Жүйке жүйесінің мүшелерінің ПРе- және ПОстнатальды дамуы. Адам онтогенезінде функционалды жүйелер туралы түсінік Белки. Химические свойства белков
Белки. Химические свойства белков Дубильні речовини. Класифiкацiя
Дубильні речовини. Класифiкацiя Вітаміни на підвіконні
Вітаміни на підвіконні Гигиена зрения. Предупреждение глазных заболеваний.
Гигиена зрения. Предупреждение глазных заболеваний. Митоз. Митоздық цикл
Митоз. Митоздық цикл Обучение животных языку. Эксперименты
Обучение животных языку. Эксперименты Синтетическая теория эволюции
Синтетическая теория эволюции Свойства и функции белков
Свойства и функции белков Ядовитые животные
Ядовитые животные Птахи нашої місцевості
Птахи нашої місцевості Николай Иванович Вавилов
Николай Иванович Вавилов Механизм мышечного сокращения
Механизм мышечного сокращения Презентация к уроку биологии: Вывих, растяжение, перелом. Первая помощь
Презентация к уроку биологии: Вывих, растяжение, перелом. Первая помощь Өсімдіктану негіздері
Өсімдіктану негіздері Биологические полимеры - нуклеиновые кислоты
Биологические полимеры - нуклеиновые кислоты Stem and system of stalk
Stem and system of stalk