Хроматографические методы анализа и их применение для контроля качества лекарственных средств (продолжение) презентация
- Хроматографические методы анализа и их применение для контроля качества лекарственных средств (продолжение)

Содержание
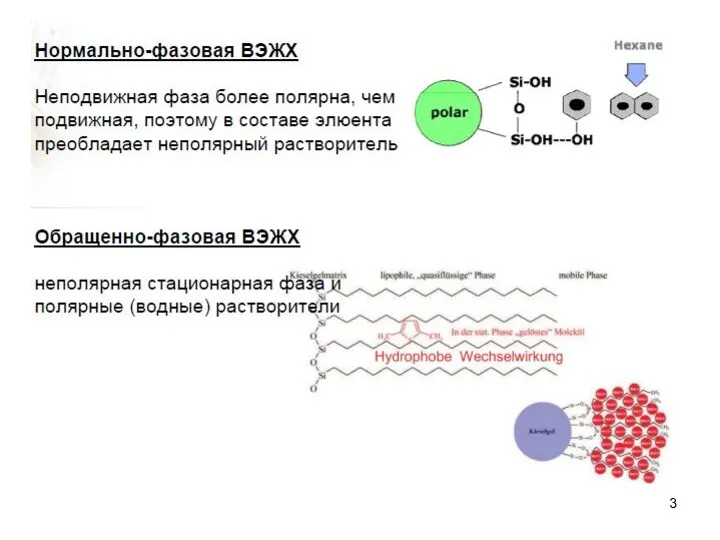
- 2. Жидкостная хроматография метод разделения, в котором подвижная фаза представляет собой жидкость, а неподвижная – твердую или
- 4. Оборудование (принципиальная схема)
- 5. Варианты проведения ЖХ 1. Изократический режим – постоянный состав ПФ в течение всего анализа (разделение родственных
- 6. Хроматографические колонки Стальные трубки внутренним диаметром 2-5 мм, длиной 5-30 см, с пористыми фильтрами с обоих
- 7. Неподвижные фазы Общие требования: 1. Для обеспечения высокой эффективности разделения – размер частиц сорбента должен быть
- 8. Неподвижные фазы Классификация: 1. Полярные фазы (для прямофазной ЖХ) – немодифицированные силикагели, аминопропилсилилсиликагели, диольные производные силилсиликагеля,
- 9. Выбор подвижной фазы для нормально-фазовой хроматографии
- 10. Неподвижные фазы 2. Среднеполярные фазы (для обращенно-фазовой ЖХ) – химически модифицированные силикагели с привитыми цианопропильными (СN),
- 11. Неподвижные фазы 3. Неполярные фазы (для обращенно-фазовой ЖХ) – химически модифицированные силикагели с привитыми октильными (С8),
- 13. Принципы разделения 1. Полярные фазы – 1.1. для разделения неполярных и малополярных веществ (слабо адсорбируются НФ)
- 14. Принципы разделения 2. Средне- и неполярные фазы: 2.1. Для разделения неполярных веществ используются ПФ с высоким
- 15. Механизмы удерживания
- 16. Закономерности удерживания в обращенно-фазовой ВЭЖХ Вытеснительная модель удерживания: 1. Из ПФ на поверхности неполярного/среднеполярного сорбента адсорбируется
- 17. Влияние рН ПФ и температуры На неполярных фазах за счет дисперсионных взаимодействий лучше удерживаются неионизированные молекулы.
- 18. Детекторы в ЖХ 1. Детектор спектрофотометрический (область длин волн – УФ 190-360 нм (дейтериевая лампа), видимая
- 19. 2. Детектор на основе диодной матрицы – сканирование оптической плотности элюата в заданном диапазоне длин волн
- 20. СФ-детектор Детектор на основе диодной матрицы
- 21. Флуориметрический детектор Селективный детектор, основанный на измерении интенсивности флуоресценции разделенных веществ (устанавливаются длины волн возбуждения и
- 22. Рефрактометрический детектор Основан на измерении величины преломления света элюата (универсальный детектор)
- 23. Электрометрические детекторы 1. Амперометрический детектор (детекция органических веществ, обладающих ОВ-свойствами) – может быть комплексный с ферментативными
- 24. Масс-спектрометрическое детектирование Универсальный (полный ионный ток) и селективный (сканирование индивидуальных масс ионов) детектор
- 26. Масс-спектрометрическое детектирование 1. Ионизация образца (ПФ + разделенные вещества) Принципы: 1.1. электрораспылительная (ESI)
- 28. Примеры ионизации
- 29. Масс-спектрометрическое детектирование 1.2. Химическая ионизация при атмосферном давлении:
- 30. 1.3.MALDI (ионизация лазерной десорбцией при взаимодействии с матрицей)
- 31. MALDI-TOF (ионизация лазерной десорбцией при взаимодействии с матрицей и времяпролетным детектированием)
- 32. Масс-анализаторы А. непрерывные масс-анализаторы 1. Магнитный и электростатический секторный масс-анализатор (Sector) 2. Квадрупольный масс-анализатор (Quadrupole mass
- 33. Масс-спектрометрическое детектирование Достоинства: 1. Высочайшая чувствительность органических веществ, биополимеров (10-15 г/пробе). 2. Высокая специфичность детекции (последовательная
- 34. Сравнение различных типов детекторов
- 35. Применение ВЭЖХ в фармацевтическом анализе 1. Идентификация веществ 1.1. Сравнение времен удерживания со стандартным веществом Раствор
- 36. Применение ВЭЖХ в фармацевтическом анализе 1.2. Идентификация по времена удерживания и спектрам поглощения пиков.
- 37. Применение ВЭЖХ в фармацевтическом анализе 1.3. Идентификация веществ (образцов) по хроматографическому профилю (наличие определенного числа пиков
- 38. Применение ВЭЖХ в фармацевтическом анализе 2. Определение специфических (родственных) примесей – полуколичественный анализ 2.1. По стандартному
- 39. 2. Определение специфических (родственных) примесей – полуколичественный анализ 2.2. По стандартному раствору основного вещества, разведенному до
- 40. 2. Определение специфических (родственных) примесей – полуколичественный анализ 2.3. Методом внутренней нормализации
- 41. Определение специфических (родственных) примесей 2.4. Количественное определение (например, токсичные примеси) – методом градуировочного графика
- 42. 2.5. Определение энантиомерной чистоты
- 43. Применение ВЭЖХ в фармацевтическом анализе 3. Определение основных показателей готовых лекарственных средств – однородность дозированных единиц,
- 44. Капиллярный электрофорез Метод капиллярного электрофореза (КЭ) основан на разделении заряженных компонентов сложной смеси в кварцевом капилляре
- 45. Капиллярный электрофорез МЭКХ - вариант капиллярного электрофореза, который позволяет проводить разделение соединений ионного и нейтрального характера
- 46. После подачи к концам капилляра высокого напряжения (до 30 кВ), компоненты смеси начинают двигаться по капилляру
- 47. Основные параметры КЭ 1. Время миграции (tм) - время, необходимое компоненту для прохождения им эффективной длины
- 49. Механизм ЭОП
- 50. Электроосмотический поток Уникальной особенностью ЭОП является плоский профиль потока в капилляре. Такой профиль выгоден, поскольку уменьшается
- 51. Капилляры для разделения Подавляющее большинство разделений в КЭ проводят с использованием кварцевых капилляров имеющих внешнее полимерное
- 52. Ввод образца Проба может быть введена в капилляр электрофоретическим, электрокинетическим или вытеснительным способом. Объем вводимой пробы
- 53. Детектирование
- 54. Пример разделения неорганических ионов (КЭ) 1 – хлорид; 2 – нитрит; 3 – сульфат; 4 –
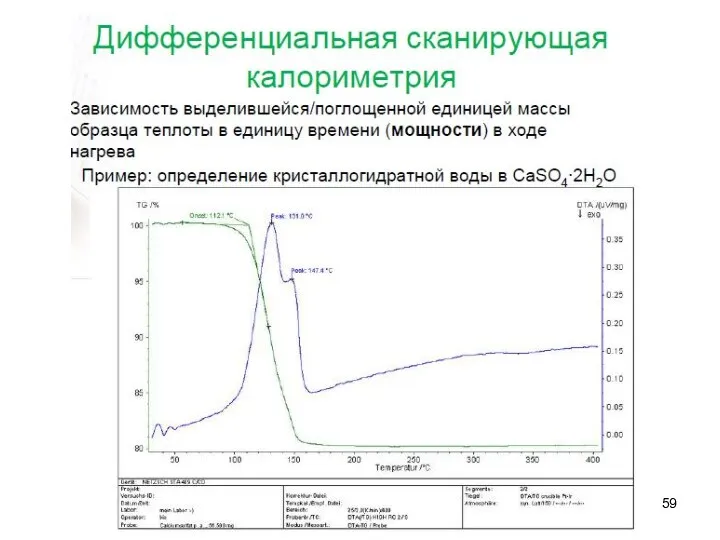
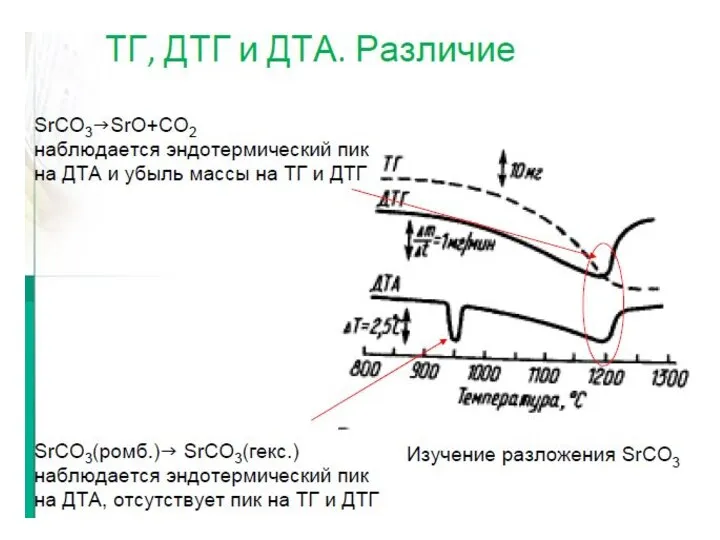
- 56. Термические методы анализа Основаны на установлении зависимостей различных физических или физико-химических свойств веществ от температуры (градиента
- 70. Скачать презентацию

Жидкостная хроматография
метод разделения, в котором подвижная фаза представляет собой жидкость, а
Жидкостная хроматография
метод разделения, в котором подвижная фаза представляет собой жидкость, а
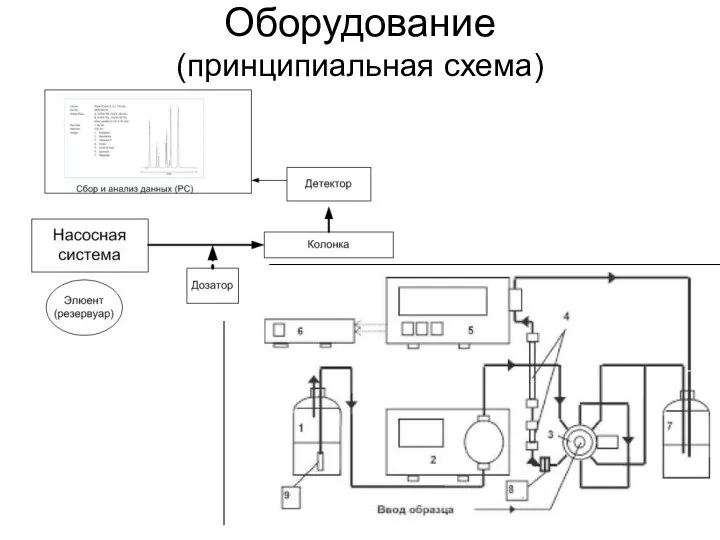
Оборудование
(принципиальная схема)
Оборудование
(принципиальная схема)
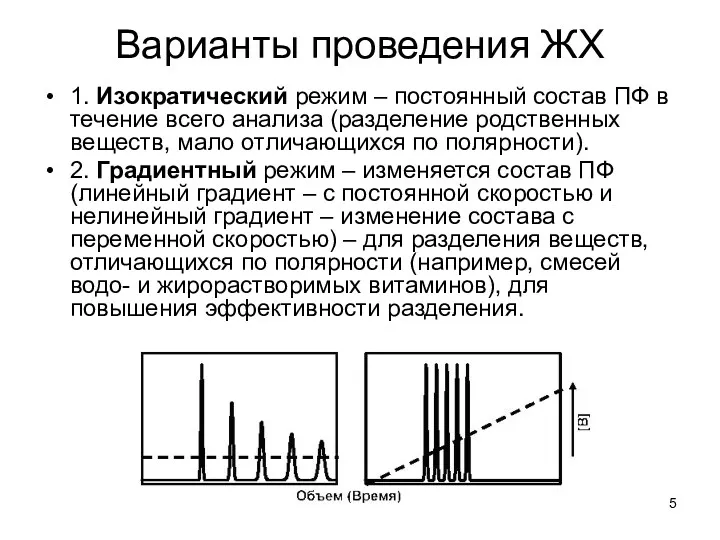
Варианты проведения ЖХ
1. Изократический режим – постоянный состав ПФ в течение
Варианты проведения ЖХ
1. Изократический режим – постоянный состав ПФ в течение

Хроматографические колонки
Стальные трубки внутренним диаметром 2-5 мм, длиной 5-30 см,
Хроматографические колонки
Стальные трубки внутренним диаметром 2-5 мм, длиной 5-30 см,

Неподвижные фазы
Общие требования:
1. Для обеспечения высокой эффективности разделения – размер частиц
Неподвижные фазы
Общие требования:
1. Для обеспечения высокой эффективности разделения – размер частиц
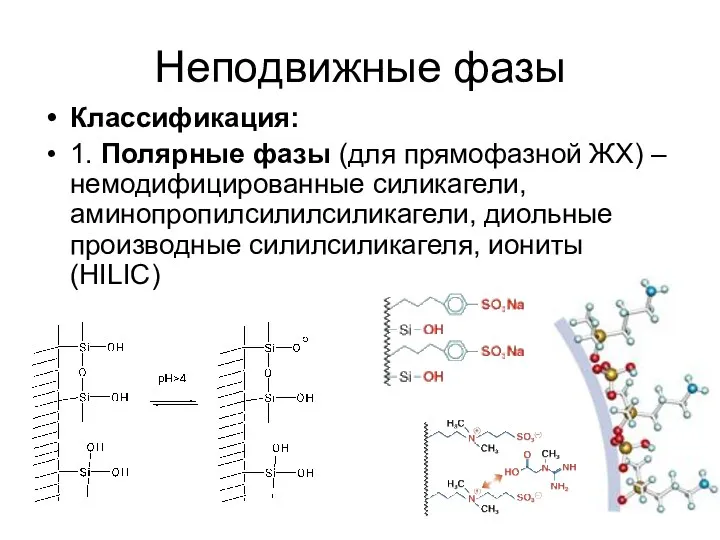
Неподвижные фазы
Классификация:
1. Полярные фазы (для прямофазной ЖХ) – немодифицированные силикагели, аминопропилсилилсиликагели,
Неподвижные фазы
Классификация:
1. Полярные фазы (для прямофазной ЖХ) – немодифицированные силикагели, аминопропилсилилсиликагели,
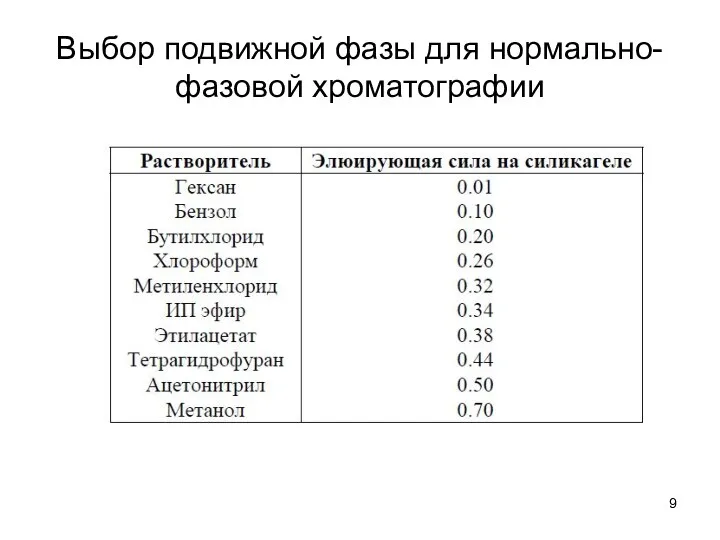
Выбор подвижной фазы для нормально-фазовой хроматографии
Выбор подвижной фазы для нормально-фазовой хроматографии
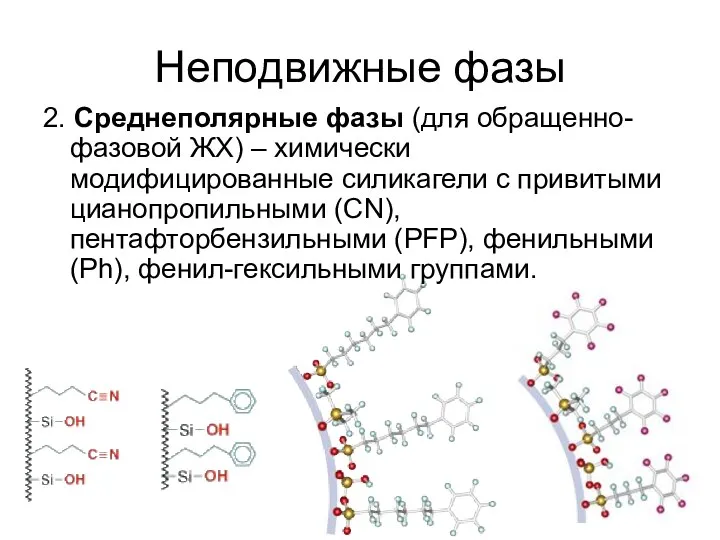
Неподвижные фазы
2. Среднеполярные фазы (для обращенно-фазовой ЖХ) – химически модифицированные силикагели
Неподвижные фазы
2. Среднеполярные фазы (для обращенно-фазовой ЖХ) – химически модифицированные силикагели

Неподвижные фазы
3. Неполярные фазы (для обращенно-фазовой ЖХ) – химически модифицированные силикагели
Неподвижные фазы
3. Неполярные фазы (для обращенно-фазовой ЖХ) – химически модифицированные силикагели
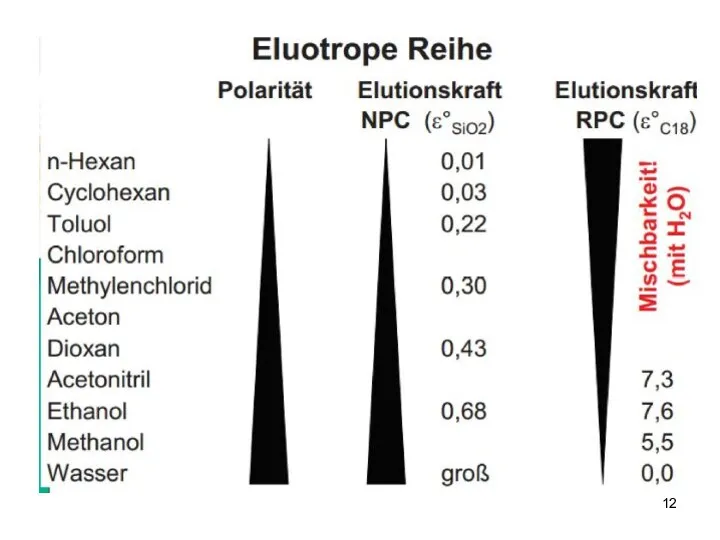

Принципы разделения
1. Полярные фазы –
1.1. для разделения неполярных и малополярных
Принципы разделения
1. Полярные фазы –
1.1. для разделения неполярных и малополярных

Принципы разделения
2. Средне- и неполярные фазы:
2.1. Для разделения неполярных веществ используются
Принципы разделения
2. Средне- и неполярные фазы:
2.1. Для разделения неполярных веществ используются
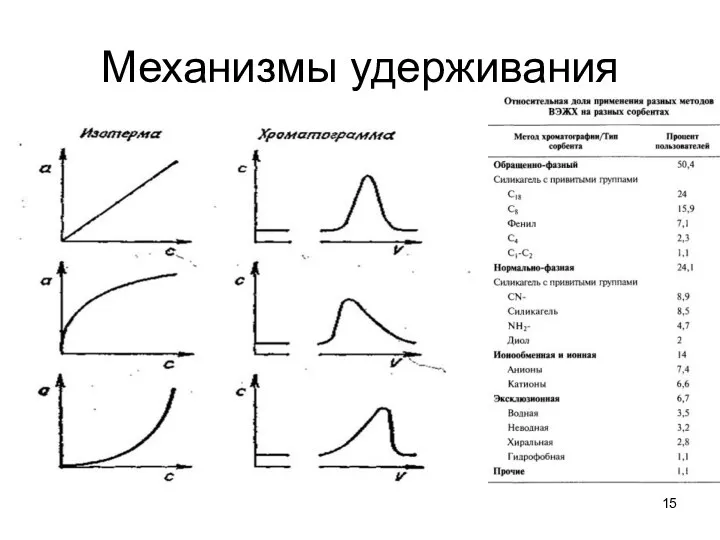
Механизмы удерживания
Механизмы удерживания
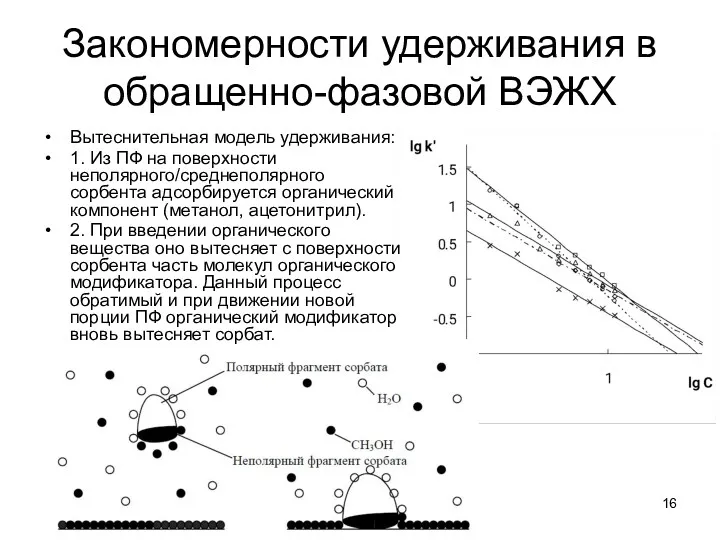
Закономерности удерживания в обращенно-фазовой ВЭЖХ
Вытеснительная модель удерживания:
1. Из ПФ на
Закономерности удерживания в обращенно-фазовой ВЭЖХ
Вытеснительная модель удерживания:
1. Из ПФ на

Влияние рН ПФ и температуры
На неполярных фазах за счет дисперсионных взаимодействий
Влияние рН ПФ и температуры
На неполярных фазах за счет дисперсионных взаимодействий
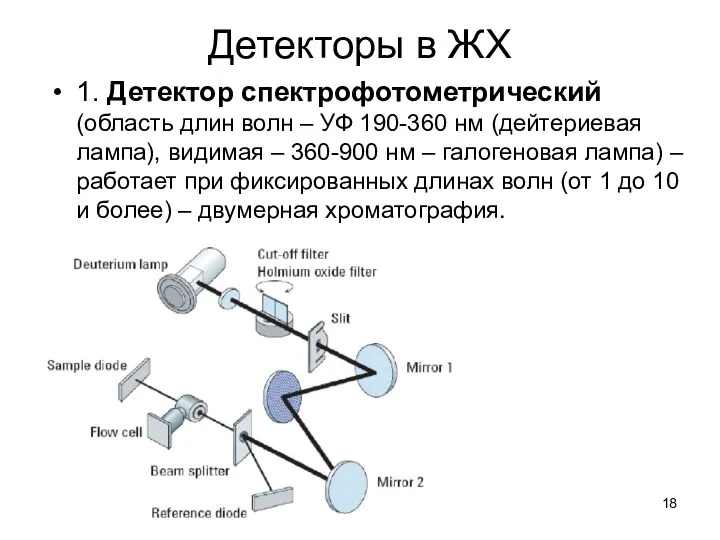
Детекторы в ЖХ
1. Детектор спектрофотометрический (область длин волн – УФ 190-360
Детекторы в ЖХ
1. Детектор спектрофотометрический (область длин волн – УФ 190-360
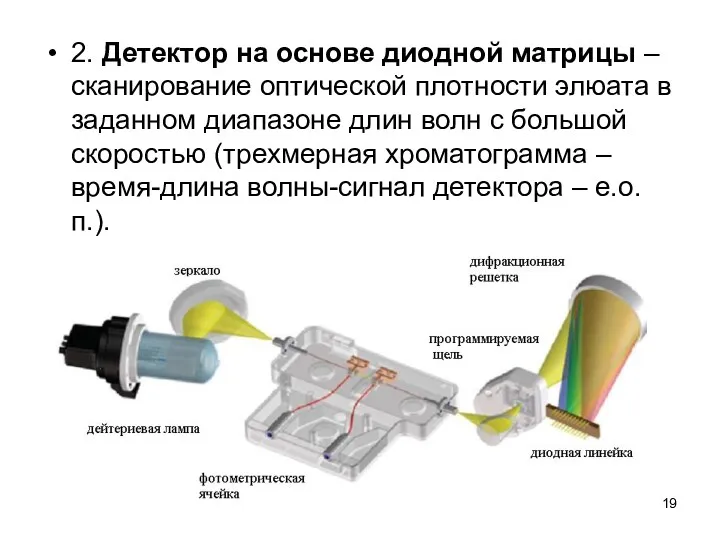
2. Детектор на основе диодной матрицы – сканирование оптической плотности элюата
2. Детектор на основе диодной матрицы – сканирование оптической плотности элюата
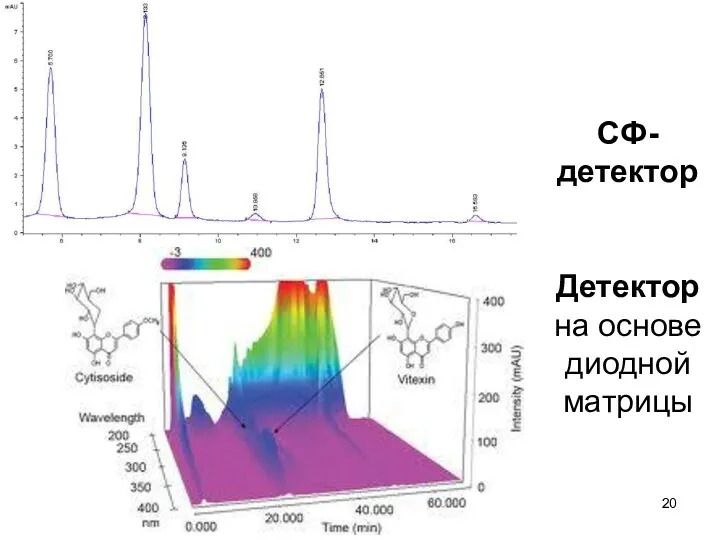
СФ-детектор
Детектор на основе диодной матрицы
СФ-детектор
Детектор на основе диодной матрицы
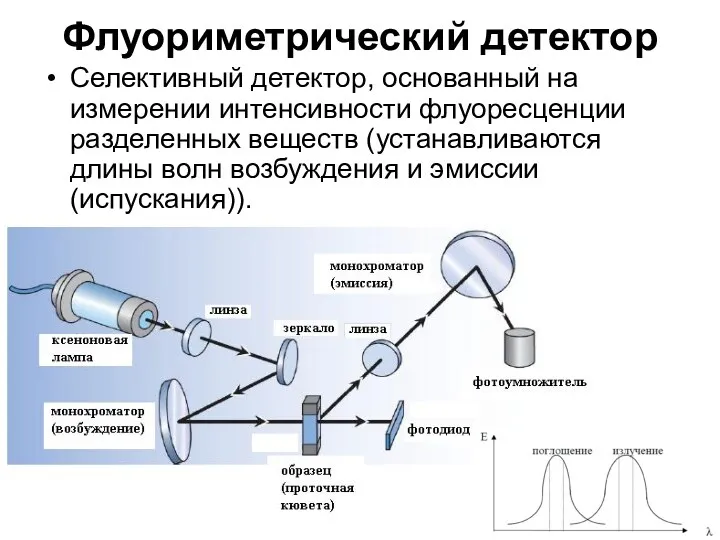
Флуориметрический детектор
Селективный детектор, основанный на измерении интенсивности флуоресценции разделенных веществ (устанавливаются
Флуориметрический детектор
Селективный детектор, основанный на измерении интенсивности флуоресценции разделенных веществ (устанавливаются
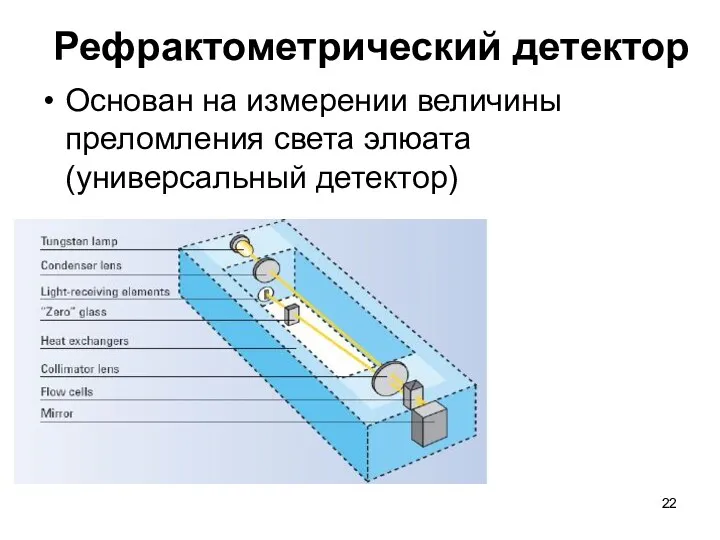
Рефрактометрический детектор
Основан на измерении величины преломления света элюата (универсальный детектор)
Рефрактометрический детектор
Основан на измерении величины преломления света элюата (универсальный детектор)
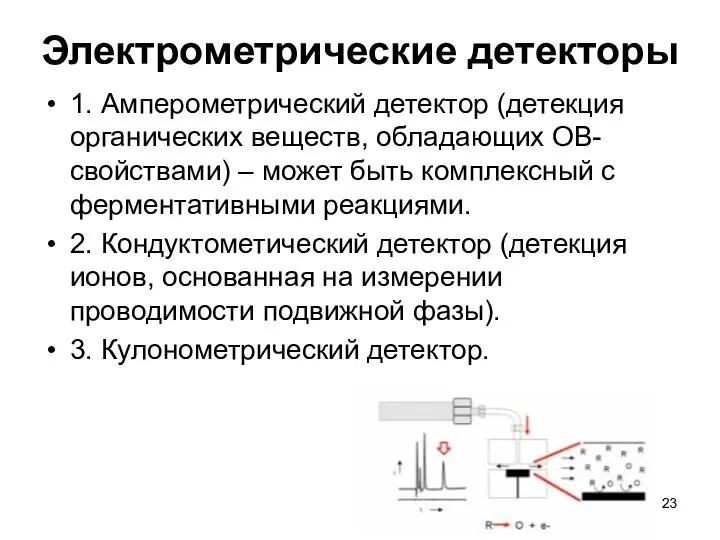
Электрометрические детекторы
1. Амперометрический детектор (детекция органических веществ, обладающих ОВ-свойствами) – может
Электрометрические детекторы
1. Амперометрический детектор (детекция органических веществ, обладающих ОВ-свойствами) – может
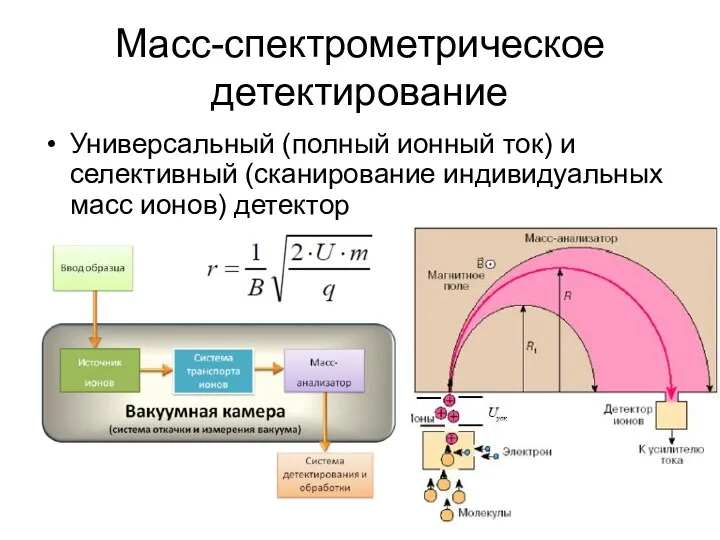
Масс-спектрометрическое детектирование
Универсальный (полный ионный ток) и селективный (сканирование индивидуальных масс ионов)
Масс-спектрометрическое детектирование
Универсальный (полный ионный ток) и селективный (сканирование индивидуальных масс ионов)

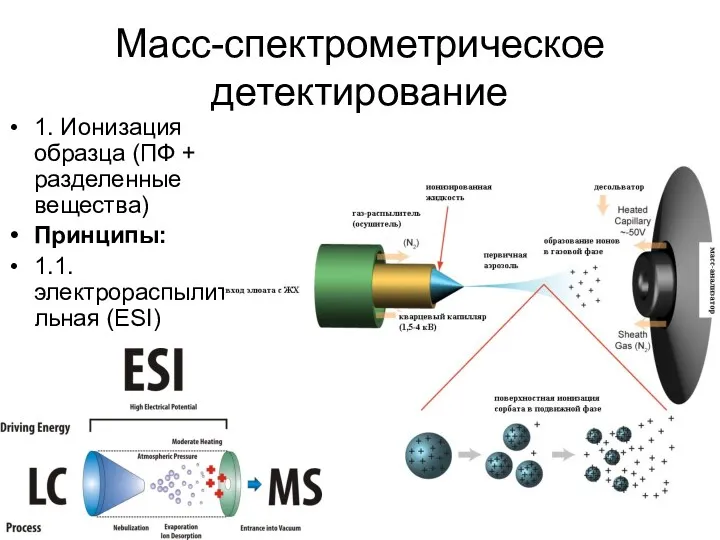
Масс-спектрометрическое детектирование
1. Ионизация образца (ПФ + разделенные вещества)
Принципы:
1.1. электрораспылительная (ESI)
Масс-спектрометрическое детектирование
1. Ионизация образца (ПФ + разделенные вещества)
Принципы:
1.1. электрораспылительная (ESI)
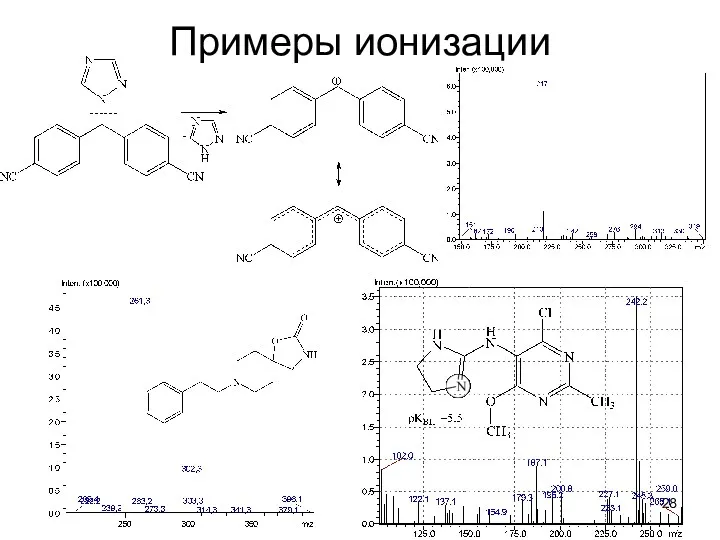
Примеры ионизации
Примеры ионизации
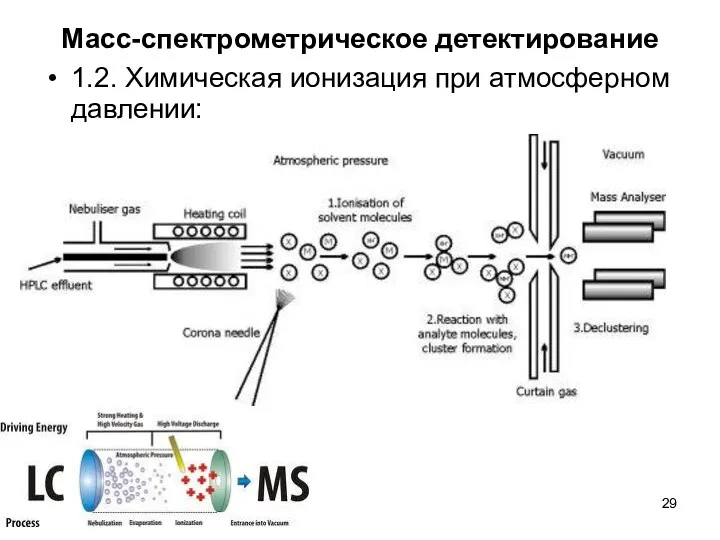
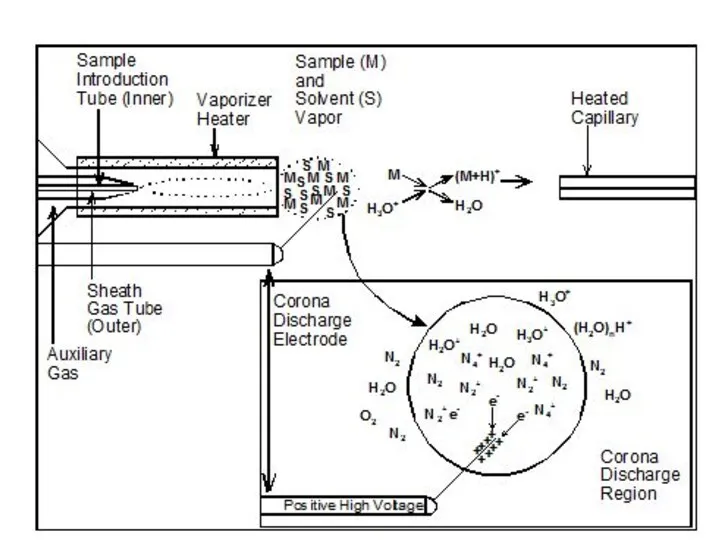
Масс-спектрометрическое детектирование
1.2. Химическая ионизация при атмосферном давлении:
Масс-спектрометрическое детектирование
1.2. Химическая ионизация при атмосферном давлении:
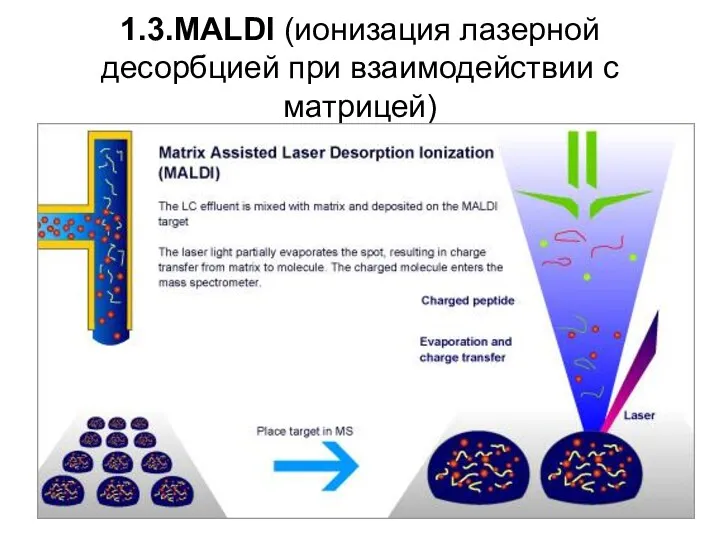
1.3.MALDI (ионизация лазерной десорбцией при взаимодействии с матрицей)
1.3.MALDI (ионизация лазерной десорбцией при взаимодействии с матрицей)
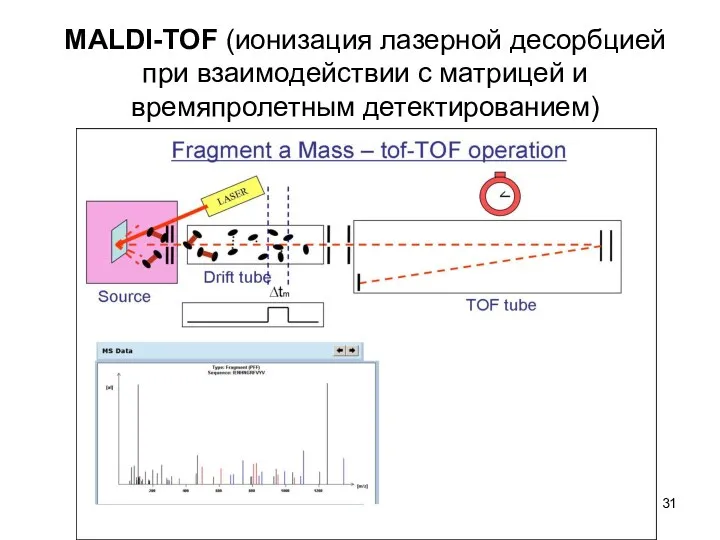
MALDI-TOF (ионизация лазерной десорбцией при взаимодействии с матрицей и времяпролетным детектированием)
MALDI-TOF (ионизация лазерной десорбцией при взаимодействии с матрицей и времяпролетным детектированием)

Масс-анализаторы
А. непрерывные масс-анализаторы
1. Магнитный и электростатический секторный масс-анализатор (Sector)
2. Квадрупольный масс-анализатор (Quadrupole mass
Масс-анализаторы
А. непрерывные масс-анализаторы
1. Магнитный и электростатический секторный масс-анализатор (Sector)
2. Квадрупольный масс-анализатор (Quadrupole mass

Масс-спектрометрическое детектирование
Достоинства:
1. Высочайшая чувствительность органических веществ, биополимеров (10-15 г/пробе).
2. Высокая специфичность
Масс-спектрометрическое детектирование
Достоинства:
1. Высочайшая чувствительность органических веществ, биополимеров (10-15 г/пробе).
2. Высокая специфичность
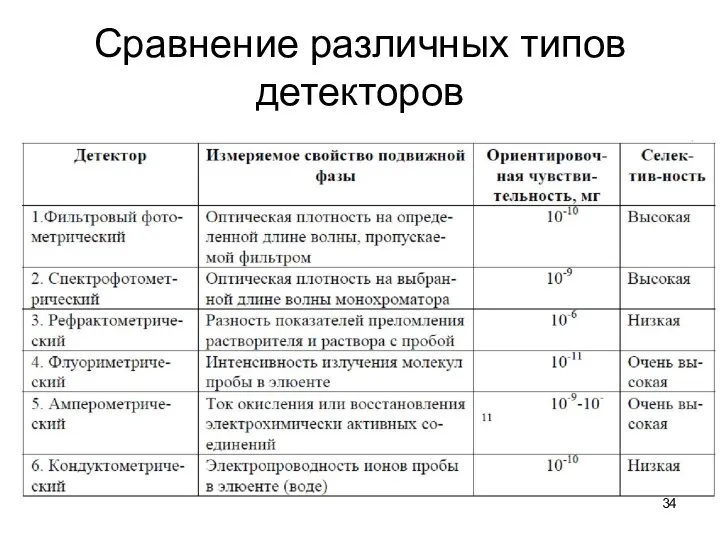
Сравнение различных типов детекторов
Сравнение различных типов детекторов
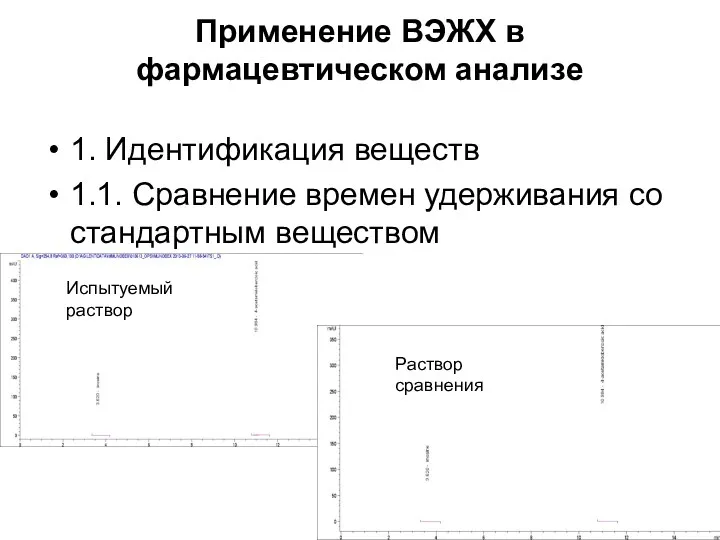
Применение ВЭЖХ в фармацевтическом анализе
1. Идентификация веществ
1.1. Сравнение времен удерживания со
Применение ВЭЖХ в фармацевтическом анализе
1. Идентификация веществ
1.1. Сравнение времен удерживания со
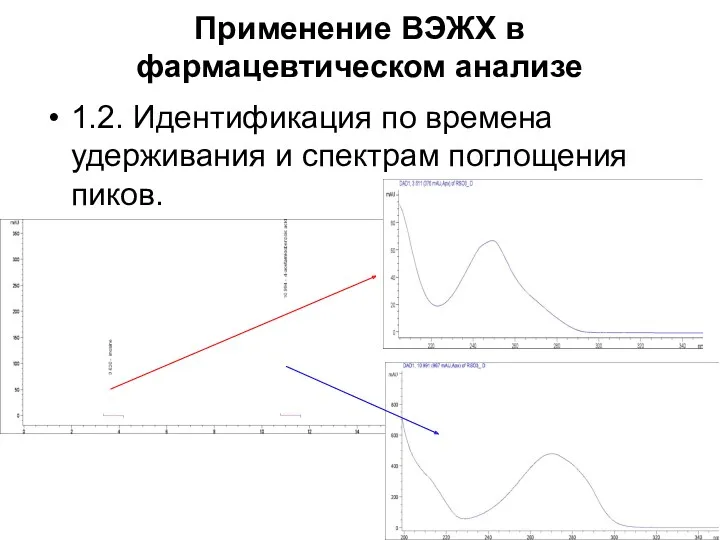
Применение ВЭЖХ в фармацевтическом анализе
1.2. Идентификация по времена удерживания и спектрам
Применение ВЭЖХ в фармацевтическом анализе
1.2. Идентификация по времена удерживания и спектрам
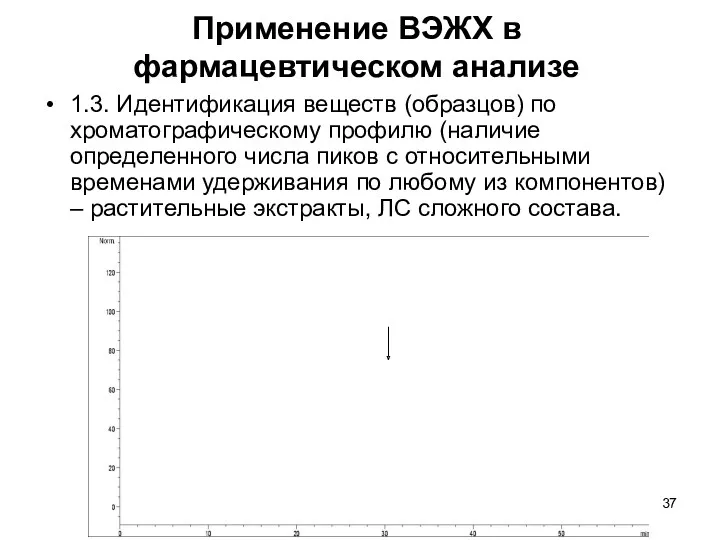
Применение ВЭЖХ в фармацевтическом анализе
1.3. Идентификация веществ (образцов) по хроматографическому профилю
Применение ВЭЖХ в фармацевтическом анализе
1.3. Идентификация веществ (образцов) по хроматографическому профилю
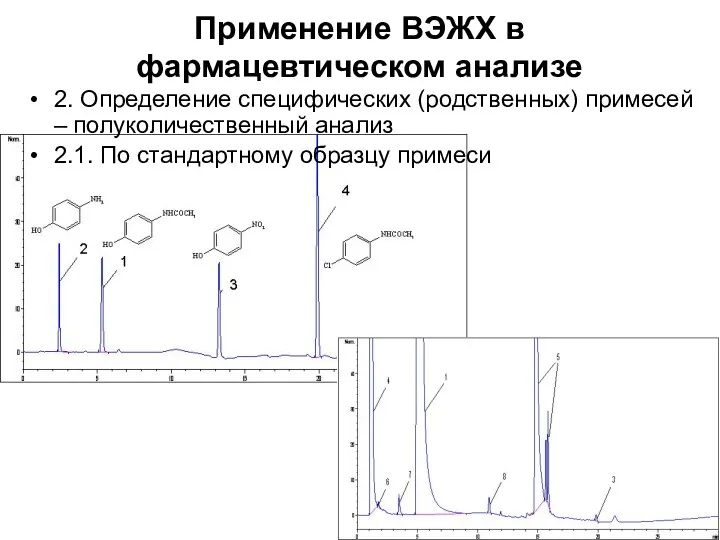
Применение ВЭЖХ в фармацевтическом анализе
2. Определение специфических (родственных) примесей – полуколичественный
Применение ВЭЖХ в фармацевтическом анализе
2. Определение специфических (родственных) примесей – полуколичественный
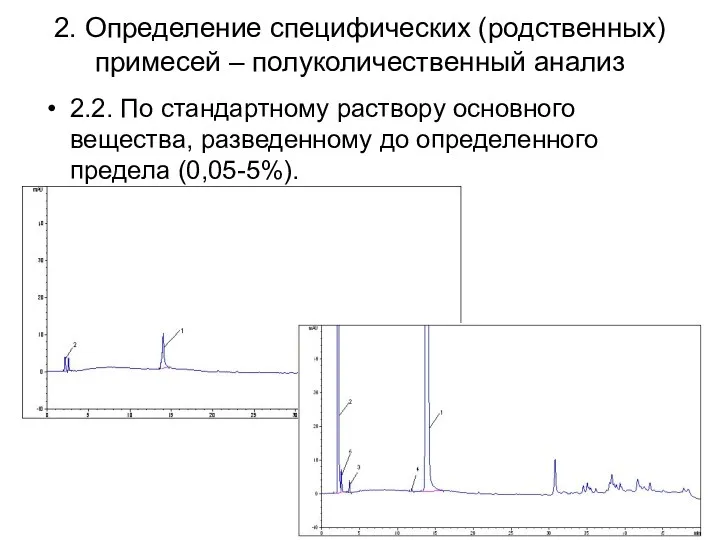
2. Определение специфических (родственных) примесей – полуколичественный анализ
2.2. По стандартному раствору
2. Определение специфических (родственных) примесей – полуколичественный анализ
2.2. По стандартному раствору
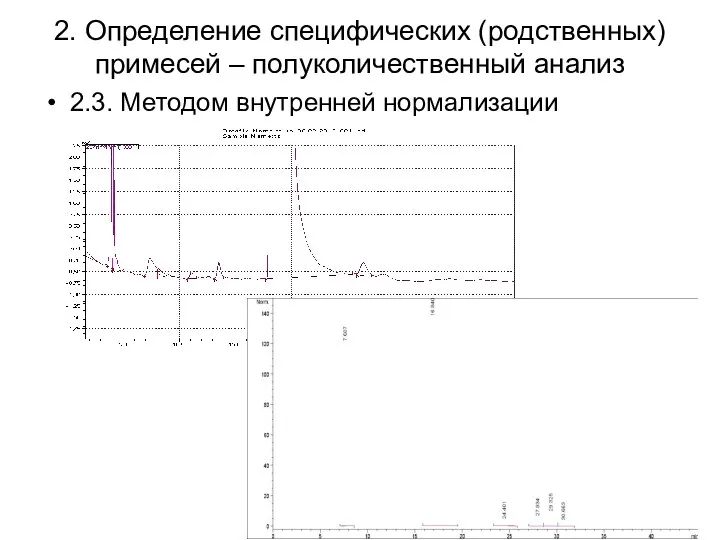
2. Определение специфических (родственных) примесей – полуколичественный анализ
2.3. Методом внутренней нормализации
2. Определение специфических (родственных) примесей – полуколичественный анализ
2.3. Методом внутренней нормализации
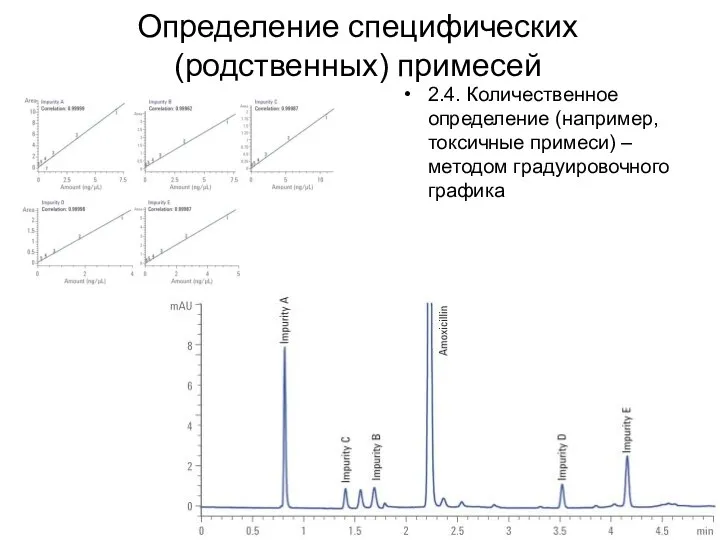
Определение специфических (родственных) примесей
2.4. Количественное определение (например, токсичные примеси) – методом
Определение специфических (родственных) примесей
2.4. Количественное определение (например, токсичные примеси) – методом
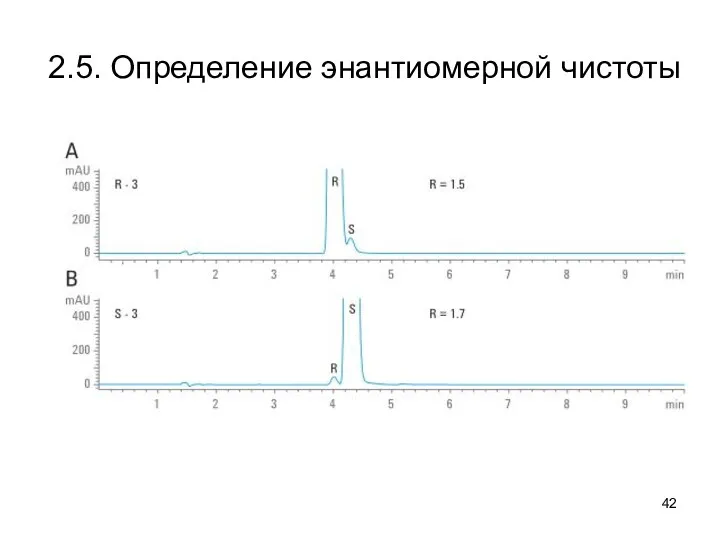
2.5. Определение энантиомерной чистоты
2.5. Определение энантиомерной чистоты

Применение ВЭЖХ в фармацевтическом анализе
3. Определение основных показателей готовых лекарственных средств
Применение ВЭЖХ в фармацевтическом анализе
3. Определение основных показателей готовых лекарственных средств

Капиллярный электрофорез
Метод капиллярного электрофореза (КЭ) основан на разделении заряженных компонентов сложной
Капиллярный электрофорез
Метод капиллярного электрофореза (КЭ) основан на разделении заряженных компонентов сложной

Капиллярный электрофорез
МЭКХ - вариант капиллярного электрофореза, который позволяет проводить разделение соединений
Капиллярный электрофорез
МЭКХ - вариант капиллярного электрофореза, который позволяет проводить разделение соединений
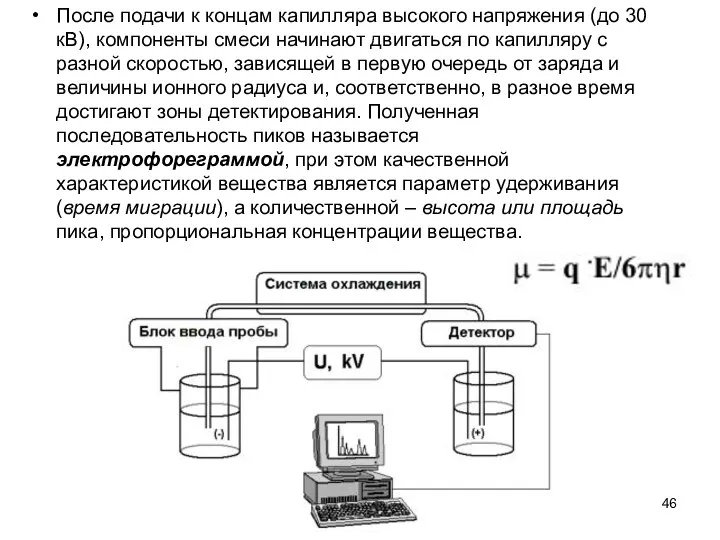
После подачи к концам капилляра высокого напряжения (до 30 кВ), компоненты
После подачи к концам капилляра высокого напряжения (до 30 кВ), компоненты

Основные параметры КЭ
1. Время миграции (tм) - время, необходимое компоненту для
Основные параметры КЭ
1. Время миграции (tм) - время, необходимое компоненту для
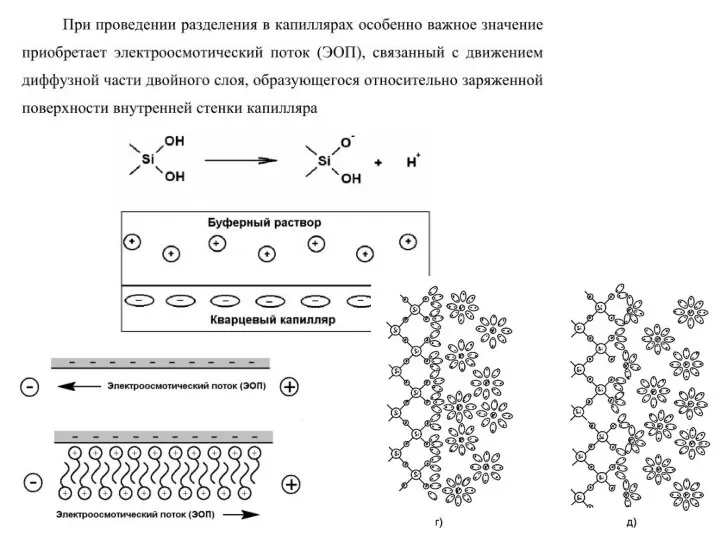
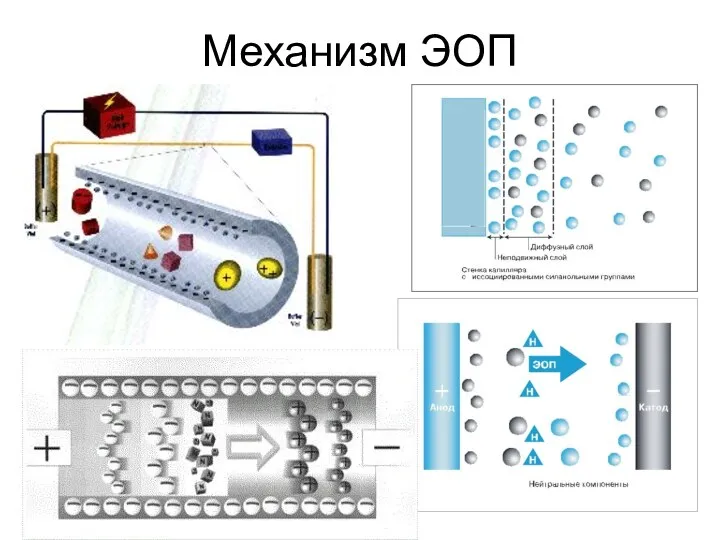
Механизм ЭОП
Механизм ЭОП

Электроосмотический поток
Уникальной особенностью ЭОП является плоский профиль потока в капилляре. Такой
Электроосмотический поток
Уникальной особенностью ЭОП является плоский профиль потока в капилляре. Такой

Капилляры для разделения
Подавляющее большинство разделений в КЭ проводят с использованием кварцевых
Капилляры для разделения
Подавляющее большинство разделений в КЭ проводят с использованием кварцевых

Ввод образца
Проба может быть введена в капилляр электрофоретическим, электрокинетическим или вытеснительным
Ввод образца
Проба может быть введена в капилляр электрофоретическим, электрокинетическим или вытеснительным

Детектирование
Детектирование
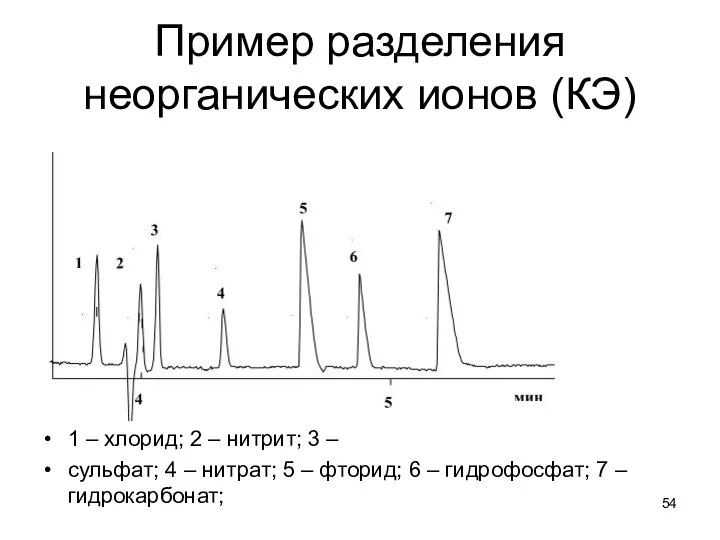
Пример разделения неорганических ионов (КЭ)
1 – хлорид; 2 – нитрит; 3
Пример разделения неорганических ионов (КЭ)
1 – хлорид; 2 – нитрит; 3


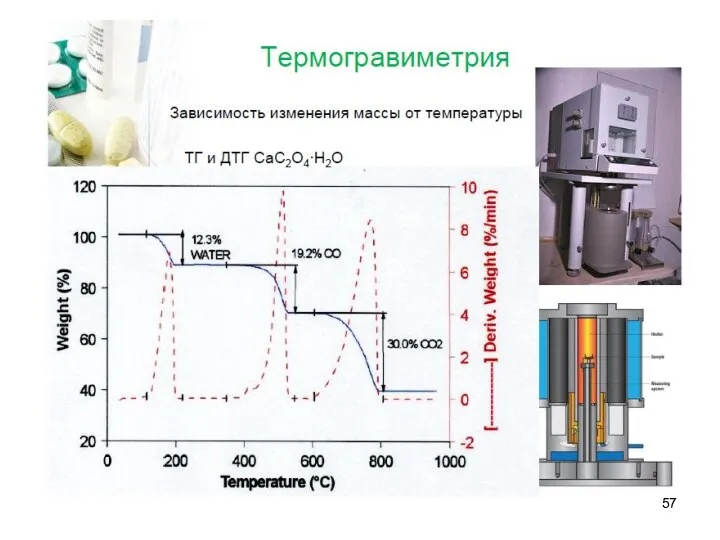
Термические методы анализа
Основаны на установлении зависимостей различных физических или физико-химических свойств
Термические методы анализа
Основаны на установлении зависимостей различных физических или физико-химических свойств








 Закон радиоактивного распада. Период полураспада
Закон радиоактивного распада. Период полураспада Ядерный магнитный резонанс
Ядерный магнитный резонанс Сила тока
Сила тока Камера-обскура. Интересные факты
Камера-обскура. Интересные факты Thermal Energy, Chemical Energy
Thermal Energy, Chemical Energy Система мащення охолодження та пуску ДВЗ
Система мащення охолодження та пуску ДВЗ ФИЗИКА 8 класс Изменение агрегатных состояний вещества
ФИЗИКА 8 класс Изменение агрегатных состояний вещества Методическое пособие по теме Геометрическая оптика
Методическое пособие по теме Геометрическая оптика Вантажопідйомні машини. Держгірпромнагляд
Вантажопідйомні машини. Держгірпромнагляд Электростатика
Электростатика Управляемый термоядерный синтез. Данные
Управляемый термоядерный синтез. Данные Люминесцентные лампы
Люминесцентные лампы Сила трения
Сила трения Жарықтың интерференциясы
Жарықтың интерференциясы Викторина на тему: Дисперсия света
Викторина на тему: Дисперсия света Рулевое управление
Рулевое управление открытый урок тепловые машины
открытый урок тепловые машины Методы и средства измерения частоты, временных интервалов и фазового сдвига
Методы и средства измерения частоты, временных интервалов и фазового сдвига Ғажайып ұяшықтар
Ғажайып ұяшықтар Семинарское занятие Основы электростатики. 10 класс.
Семинарское занятие Основы электростатики. 10 класс. Виды спектров и спектральный анализ. Тема №48
Виды спектров и спектральный анализ. Тема №48 Принципы построения системы допусков и посадок (СДП
Принципы построения системы допусков и посадок (СДП Своя игра. Простые механизмы
Своя игра. Простые механизмы Внутреннняя энергия
Внутреннняя энергия Выяснение условия равновесия рычага
Выяснение условия равновесия рычага Урок физики в 7 классе Инерция
Урок физики в 7 классе Инерция Структурный анализ плоских механизмов
Структурный анализ плоских механизмов Электричество и Магнетизм
Электричество и Магнетизм