- Хемоинформатика

Содержание
- 2. Хемоинформатика (химическая информатика, молекулярная информатика) — применение методов информатики для решения химических проблем.
- 3. Компьютерная химия (математическая химия) — сравнительно молодая область химии, основанная на применении компьютерных методов и дискретной
- 4. Хемоинформатика это научная дисциплина, возникшая за последние 40 лет в пограничной области между химией и вычислительной
- 5. Хемоинформатика, наряду с квантовой химией и молекулярным моделированием, является ветвью теоретической химии (theoretical chemistry) и областью
- 6. Компьютерное представление химической информации. Создание и управление базами данных по химии. Молекулярный дизайн химических соединений с
- 7. В хемоинформатике для внутреннего представления структур химических соединений обычно используются молекулярные графы, которые могут быть при
- 8. SMILES (Simplified Molecular Input Line Entry Specification, англ. спецификация упрощенного представления молекул в строке ввода) —
- 9. Особенностью управления базами данных по химии является то, что оно обеспечивает следующие виды поиска, характерные для
- 10. Структура Маркуша – это родовая или общая структура, объединяющая группу химических соединений, которые по определению должны
- 11. Понятие молекулярного подобия (или химического подобия, chemical similarity) является одной из ключевых концепций хемоинформатики. Оно играет
- 12. ISIS/Host, ISIS/Base (www.mdli.com) ChemFinder, ChemOffice (www.cambridgesoft.com) JChem (www.chemaxon.com) THOR (www.daylight.com) MOE (www.chemcomp.com) ICM Pro (под mySQL)
- 13. PubChem (pubchem.ncbi.nlm.nih.gov) ZINC (zinc.docking.org) NCI (129.43.27.140/ncidb2) DrugBank (www.drugbank.ca) BindingDB (www.bindingdb.org) DUD (dud.docking.org) ChemSpider (www.chemspider.com) ChEMBL (www.ebi.ac.uk)
- 14. Одной из важнейших задач хемоинформатики является молекулярный дизайн химических соединений с заданными свойствами. Под этим понимается
- 15. Одной из центральных задач хемоинформатики является визуализация и составление карт химического пространства, навигация и выявление неисследованных
- 16. Фармакофор (от др.-греч. φάρμακον «лекарство» и φορός «несущий») — это набор пространственных и электронных признаков, необходимых
- 17. Виртуальный скрининг — это вычислительная процедура, которая включает автоматизированный просмотр базы данных химических соединений и отбор
- 18. VSDocker (http://bio.nnov.ru/projects/vsdocker2) DOVIS (http://www.bhsai.org/)
- 19. Лиганд (от лат. ligare — связывать) — атом, ион или молекула, связанные с неким центром (акцептором).
- 20. Молекулярный докинг (или молекулярная стыковка) — это метод молекулярного моделирования, который позволяет предсказать наиболее выгодную для
- 21. FlexX (http://www.biosolveit.de/FlexX/) Dock (http://dock.compbio.ucsf.edu) AutoDock (http://autodock.scripps.edu) AutoDock Vina (http://vina.scripps.edu) Surflex (http://www.biopharmics.com, www.tripos.com) Fred (http://www.eyesopen.com/products/applications/fred.html) Gold (http://www.ccdc.cam.ac.uk/products/life_sciences/gold/)
- 22. АМРА-рецептор (рецептор α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты, AMPAR) — ионотропный рецептор глутамата, который передаёт быстрые возбуждающие сигналы в синапсах
- 23. NMDA-рецептор (NMDAR; НМДА-рецептор) — ионотропный рецептор глутамата, селективно связывающий N-метил-D-аспартат (NMDA). Структурно NMDA-рецептор представляет собой гетеротетрамер
- 24. Компьютерный синтез (англ. Computer Assisted Synthesis Design) — область хемоинформатики, охватывающая методы, алгоритмы и реализующие их
- 25. Ретросинтетический анализ. Синтез "вперед". Дизайн новых типов органических реакций.
- 26. Эмпирический подход к компьютерному синтезу. Неэмпирический компьютерный синтез.
- 27. EROS (Elaboration of Reactions for Organic Synthesis) TOSCA (Topological Synthesis design by Computer Application) FLAMINCOES (Formal-Logical
- 28. LHASA (Logic and Heuristic Applied to Synthetic Analysis) SECS (Simulation and Evaluation of Chemical Synthesis) REACT
- 29. Синтез "вперед" предсказывает результат органических реакций для заданных исходных веществ, реагентов и условий проведения реакций. Предсказания
- 30. • CAMEO (Computer Assisted Mechanistic Evaluation of Organic reactions) • ICAR
- 31. Компьютерные программы, предназначенные для дизайна новых типов органических реакций путём формального перечисления различных способов перераспределения связей:
- 32. Поиск количественных соотношений структура-свойство — процедура построения моделей, позволяющих по структурам химических соединений предсказывать их разнообразные
- 33. При векторном описании химической структуре ставится в соответствие вектор молекулярных дескрипторов, каждый из которых представляет собой
- 34. Существующие наборы молекулярных дескрипторов могут быть условно разделены на следующие категории: Фрагментные дескрипторы существуют в двух
- 35. Для решения регрессионных задач при векторном описании структур химических соединений чаще всего в хемоинформатике применяются следующие
- 36. Для решения одноклассовых классификационных задач при векторном описании структур химических соединений чаще всего в хемоинформатике применяются
- 37. Моделирование свойств при невекторном описании химических соединений осуществляется либо при помощи нейронных сетей специальных архитектур, позволяющих
- 38. Матрица смежности графа G с конечным числом вершин n (пронумерованных числами от 1 до n) —
- 39. Метод опорных векторов (англ. SVM, support vector machine) — набор схожих алгоритмов обучения с учителем, использующихся
- 40. Регрессия (лат. regressio — обратное движение, отход) в теории вероятностей и математической статистике — математическое выражение,
- 41. Random forest (с англ. — «случайный лес») — алгоритм машинного обучения, предложенный Лео Брейманом и Адель
- 42. Линейный дискриминантный анализ (ЛДА) - это метод поиска линейной комбинации переменных, наилучшим образом разделяющей два или
- 43. Искусственная нейронная сеть (ИНС) — математическая модель, а также её программное или аппаратное воплощение, построенная по
- 44. Примерами служащих для этой цели графовых (либо химических, фармакофорных) ядер являются: 1. Marginalized graph kernel 2.
- 45. Метод k ближайших соседей (англ. k-nearest neighbors algorithm, k-NN) — метрический алгоритм для автоматической классификации объектов.
- 46. Физические свойства индивидуальных низкомолекулярных соединений Температура кипения (Тк) Критическая температура (Tкр) Вязкость Давление насыщенного пара Плотность
- 47. Спектроскопические свойства Положение длинноволновой полосы поглощения симметричных цианиновых красителей Химические сдвиги в спектрах 1H ЯМР Химические
- 48. Физические свойства, обусловленные межмолекулярными взаимодействиями молекул разного типа 1. Растворимость в воде (LogSw) 2. Коэффициент распределения
- 50. Супрамолекулярные свойства Стабильность комплексов включения органических соединений с бета-циклодекстрином Сродство красителей к целлюлозному волокну Константы устойчивости
- 51. Физические и физико-химические свойства полимеров Температура стеклования Показатель преломления полимеров Ускорение вулканизации резин Коэффициент проницаемости через
- 52. Физическо-химические свойства низкомолекулярных соединений Температура вспышки и температура самовоспламенения Октановые числа углеводородов Константы ионизации (кислотности или
- 53. Фармакокинетические свойства Проникновение через гематоэнцефалический барьер Скорость проникновения через кожу Метаболизм Сайты ароматического гидроксилирования при метаболической
- 54. Ресурсы, позволяющие строить новые модели структура-свойство: Online CHemical Modeling (OCHEM) — информационный и вычислительный ресурс, позволяющий
- 56. Скачать презентацию
Хемоинформатика (химическая информатика, молекулярная информатика) — применение методов информатики для решения
Хемоинформатика (химическая информатика, молекулярная информатика) — применение методов информатики для решения
Компьютерная химия (математическая химия) — сравнительно молодая область химии, основанная на
Компьютерная химия (математическая химия) — сравнительно молодая область химии, основанная на

Хемоинформатика это научная дисциплина, возникшая за последние 40 лет в пограничной
Хемоинформатика это научная дисциплина, возникшая за последние 40 лет в пограничной

Хемоинформатика, наряду с квантовой химией и молекулярным моделированием, является ветвью теоретической
Хемоинформатика, наряду с квантовой химией и молекулярным моделированием, является ветвью теоретической
Компьютерное представление химической информации.
Создание и управление базами данных по химии.
Молекулярный дизайн
Компьютерное представление химической информации.
Создание и управление базами данных по химии.
Молекулярный дизайн

В хемоинформатике для внутреннего представления структур химических соединений обычно используются молекулярные
В хемоинформатике для внутреннего представления структур химических соединений обычно используются молекулярные

SMILES (Simplified Molecular Input Line Entry Specification, англ. спецификация упрощенного представления
SMILES (Simplified Molecular Input Line Entry Specification, англ. спецификация упрощенного представления

Особенностью управления базами данных по химии является то, что оно обеспечивает
Особенностью управления базами данных по химии является то, что оно обеспечивает

Структура Маркуша – это родовая или общая структура, объединяющая группу химических
Структура Маркуша – это родовая или общая структура, объединяющая группу химических

Понятие молекулярного подобия (или химического подобия, chemical similarity) является одной из
Понятие молекулярного подобия (или химического подобия, chemical similarity) является одной из
ISIS/Host, ISIS/Base (www.mdli.com)
ChemFinder, ChemOffice (www.cambridgesoft.com)
JChem (www.chemaxon.com)
THOR (www.daylight.com)
MOE (www.chemcomp.com)
ICM Pro (под mySQL)
ISIS/Host, ISIS/Base (www.mdli.com)
ChemFinder, ChemOffice (www.cambridgesoft.com)
JChem (www.chemaxon.com)
THOR (www.daylight.com)
MOE (www.chemcomp.com)
ICM Pro (под mySQL)

PubChem (pubchem.ncbi.nlm.nih.gov)
ZINC (zinc.docking.org)
NCI (129.43.27.140/ncidb2)
DrugBank (www.drugbank.ca)
BindingDB (www.bindingdb.org)
DUD (dud.docking.org)
ChemSpider (www.chemspider.com)
ChEMBL (www.ebi.ac.uk)
ChEBI (www.ebi.ac.uk)
PubChem (pubchem.ncbi.nlm.nih.gov)
ZINC (zinc.docking.org)
NCI (129.43.27.140/ncidb2)
DrugBank (www.drugbank.ca)
BindingDB (www.bindingdb.org)
DUD (dud.docking.org)
ChemSpider (www.chemspider.com)
ChEMBL (www.ebi.ac.uk)
ChEBI (www.ebi.ac.uk)

Одной из важнейших задач хемоинформатики является молекулярный дизайн химических соединений с
Одной из важнейших задач хемоинформатики является молекулярный дизайн химических соединений с

Одной из центральных задач хемоинформатики является визуализация и составление карт химического
Одной из центральных задач хемоинформатики является визуализация и составление карт химического

Фармакофор (от др.-греч. φάρμακον «лекарство» и φορός «несущий») — это набор
Фармакофор (от др.-греч. φάρμακον «лекарство» и φορός «несущий») — это набор

Виртуальный скрининг — это вычислительная процедура, которая включает автоматизированный просмотр базы
Виртуальный скрининг — это вычислительная процедура, которая включает автоматизированный просмотр базы

VSDocker (http://bio.nnov.ru/projects/vsdocker2)
DOVIS (http://www.bhsai.org/)
VSDocker (http://bio.nnov.ru/projects/vsdocker2)
DOVIS (http://www.bhsai.org/)

Лиганд (от лат. ligare — связывать) — атом, ион или молекула,
Лиганд (от лат. ligare — связывать) — атом, ион или молекула,

Молекулярный докинг (или молекулярная стыковка) — это метод молекулярного моделирования, который
Молекулярный докинг (или молекулярная стыковка) — это метод молекулярного моделирования, который

FlexX (http://www.biosolveit.de/FlexX/)
Dock (http://dock.compbio.ucsf.edu)
AutoDock (http://autodock.scripps.edu)
AutoDock Vina (http://vina.scripps.edu)
Surflex (http://www.biopharmics.com, www.tripos.com)
Fred (http://www.eyesopen.com/products/applications/fred.html)
Gold (http://www.ccdc.cam.ac.uk/products/life_sciences/gold/)
PLANTS (http://www.tcd.uni-konstanz.de/research/plants.php)
3DPL
FlexX (http://www.biosolveit.de/FlexX/)
Dock (http://dock.compbio.ucsf.edu)
AutoDock (http://autodock.scripps.edu)
AutoDock Vina (http://vina.scripps.edu)
Surflex (http://www.biopharmics.com, www.tripos.com)
Fred (http://www.eyesopen.com/products/applications/fred.html)
Gold (http://www.ccdc.cam.ac.uk/products/life_sciences/gold/)
PLANTS (http://www.tcd.uni-konstanz.de/research/plants.php)
3DPL
АМРА-рецептор (рецептор α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты, AMPAR) — ионотропный рецептор глутамата, который передаёт
АМРА-рецептор (рецептор α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты, AMPAR) — ионотропный рецептор глутамата, который передаёт
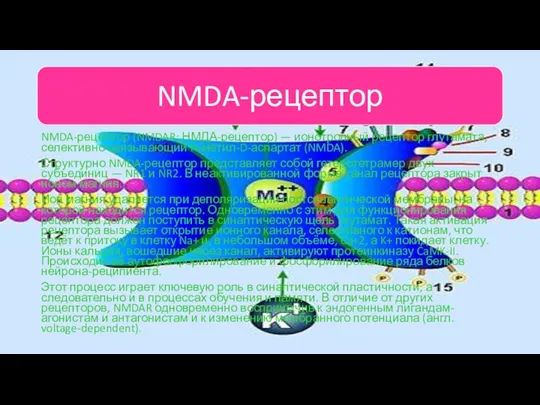
NMDA-рецептор (NMDAR; НМДА-рецептор) — ионотропный рецептор глутамата, селективно связывающий N-метил-D-аспартат (NMDA).
Структурно
NMDA-рецептор (NMDAR; НМДА-рецептор) — ионотропный рецептор глутамата, селективно связывающий N-метил-D-аспартат (NMDA).
Структурно

Компьютерный синтез (англ. Computer Assisted Synthesis Design) — область хемоинформатики, охватывающая
Компьютерный синтез (англ. Computer Assisted Synthesis Design) — область хемоинформатики, охватывающая

Ретросинтетический анализ.
Синтез "вперед".
Дизайн новых типов органических реакций.
Ретросинтетический анализ.
Синтез "вперед".
Дизайн новых типов органических реакций.

Эмпирический подход к компьютерному синтезу.
Неэмпирический компьютерный синтез.
Эмпирический подход к компьютерному синтезу.
Неэмпирический компьютерный синтез.

EROS (Elaboration of Reactions for Organic Synthesis)
TOSCA (Topological Synthesis design by Computer
EROS (Elaboration of Reactions for Organic Synthesis)
TOSCA (Topological Synthesis design by Computer

LHASA (Logic and Heuristic Applied to Synthetic Analysis)
SECS (Simulation and Evaluation of
LHASA (Logic and Heuristic Applied to Synthetic Analysis)
SECS (Simulation and Evaluation of
Синтез "вперед" предсказывает результат органических реакций для заданных исходных веществ, реагентов
Синтез "вперед" предсказывает результат органических реакций для заданных исходных веществ, реагентов

• CAMEO (Computer Assisted Mechanistic Evaluation of Organic reactions)
• ICAR
• CAMEO (Computer Assisted Mechanistic Evaluation of Organic reactions)
• ICAR

Компьютерные программы, предназначенные для дизайна новых типов органических реакций путём формального
Компьютерные программы, предназначенные для дизайна новых типов органических реакций путём формального

Поиск количественных соотношений структура-свойство — процедура построения моделей, позволяющих по структурам
Поиск количественных соотношений структура-свойство — процедура построения моделей, позволяющих по структурам

При векторном описании химической структуре ставится в соответствие вектор молекулярных дескрипторов,
При векторном описании химической структуре ставится в соответствие вектор молекулярных дескрипторов,

Существующие наборы молекулярных дескрипторов могут быть условно разделены на следующие категории:
Фрагментные
Существующие наборы молекулярных дескрипторов могут быть условно разделены на следующие категории:
Фрагментные

Для решения регрессионных задач при векторном описании структур химических соединений чаще всего в хемоинформатике применяются
Для решения регрессионных задач при векторном описании структур химических соединений чаще всего в хемоинформатике применяются

Для решения одноклассовых классификационных задач при векторном описании структур химических соединений чаще всего в хемоинформатике
Для решения одноклассовых классификационных задач при векторном описании структур химических соединений чаще всего в хемоинформатике

Моделирование свойств при невекторном описании химических соединений осуществляется либо при помощи
Моделирование свойств при невекторном описании химических соединений осуществляется либо при помощи

Матрица смежности графа G с конечным числом вершин n (пронумерованных числами
Матрица смежности графа G с конечным числом вершин n (пронумерованных числами

Метод опорных векторов (англ. SVM, support vector machine) — набор схожих
Метод опорных векторов (англ. SVM, support vector machine) — набор схожих

Регрессия (лат. regressio — обратное движение, отход) в теории вероятностей
Регрессия (лат. regressio — обратное движение, отход) в теории вероятностей

Random forest (с англ. — «случайный лес») — алгоритм машинного обучения, предложенный Лео
Random forest (с англ. — «случайный лес») — алгоритм машинного обучения, предложенный Лео

Линейный дискриминантный анализ (ЛДА) - это метод поиска линейной комбинации переменных,
Линейный дискриминантный анализ (ЛДА) - это метод поиска линейной комбинации переменных,

Искусственная нейронная сеть (ИНС) — математическая модель, а также её программное
Искусственная нейронная сеть (ИНС) — математическая модель, а также её программное

Примерами служащих для этой цели графовых (либо химических, фармакофорных) ядер являются:
1. Marginalized
Примерами служащих для этой цели графовых (либо химических, фармакофорных) ядер являются:
1. Marginalized

Метод k ближайших соседей (англ. k-nearest neighbors algorithm, k-NN) — метрический
Метод k ближайших соседей (англ. k-nearest neighbors algorithm, k-NN) — метрический

Физические свойства индивидуальных низкомолекулярных соединений
Температура кипения (Тк)
Критическая температура (Tкр)
Вязкость
Давление насыщенного пара
Плотность
Показатель преломления
Температура плавления (Тпл)
Шкалы
Физические свойства индивидуальных низкомолекулярных соединений
Температура кипения (Тк)
Критическая температура (Tкр)
Вязкость
Давление насыщенного пара
Плотность
Показатель преломления
Температура плавления (Тпл)
Шкалы

Спектроскопические свойства
Положение длинноволновой полосы поглощения симметричных цианиновых красителей
Химические сдвиги в спектрах 1H ЯМР
Химические сдвиги
Спектроскопические свойства
Положение длинноволновой полосы поглощения симметричных цианиновых красителей
Химические сдвиги в спектрах 1H ЯМР
Химические сдвиги

Физические свойства, обусловленные межмолекулярными взаимодействиями молекул разного типа
1. Растворимость в воде (LogSw)
2. Коэффициент
Физические свойства, обусловленные межмолекулярными взаимодействиями молекул разного типа
1. Растворимость в воде (LogSw)
2. Коэффициент


Супрамолекулярные свойства
Стабильность комплексов включения органических соединений с бета-циклодекстрином
Сродство красителей к целлюлозному волокну
Константы устойчивости комплексов ионофоров с ионами металлов
Физические свойства
Супрамолекулярные свойства
Стабильность комплексов включения органических соединений с бета-циклодекстрином
Сродство красителей к целлюлозному волокну
Константы устойчивости комплексов ионофоров с ионами металлов
Физические свойства

Физические и физико-химические свойства полимеров
Температура стеклования
Показатель преломления полимеров
Ускорение вулканизации резин
Коэффициент проницаемости через полиэтилен низкой плотности
Физические свойства ионных
Физические и физико-химические свойства полимеров
Температура стеклования
Показатель преломления полимеров
Ускорение вулканизации резин
Коэффициент проницаемости через полиэтилен низкой плотности
Физические свойства ионных

Физическо-химические свойства низкомолекулярных соединений
Температура вспышки и температура самовоспламенения
Октановые числа углеводородов
Константы ионизации (кислотности или основности)
Физическо-химические свойства низкомолекулярных соединений
Температура вспышки и температура самовоспламенения
Октановые числа углеводородов
Константы ионизации (кислотности или основности)

Фармакокинетические свойства
Проникновение через гематоэнцефалический барьер
Скорость проникновения через кожу
Метаболизм
Сайты ароматического гидроксилирования при метаболической
Фармакокинетические свойства
Проникновение через гематоэнцефалический барьер
Скорость проникновения через кожу
Метаболизм
Сайты ароматического гидроксилирования при метаболической

Ресурсы, позволяющие строить новые модели структура-свойство:
Online CHemical Modeling (OCHEM) — информационный и
Ресурсы, позволяющие строить новые модели структура-свойство:
Online CHemical Modeling (OCHEM) — информационный и
 Визитная карточка Свердловской городской библиотеки ЩРБИЦ Щелковского муниципального района
Визитная карточка Свердловской городской библиотеки ЩРБИЦ Щелковского муниципального района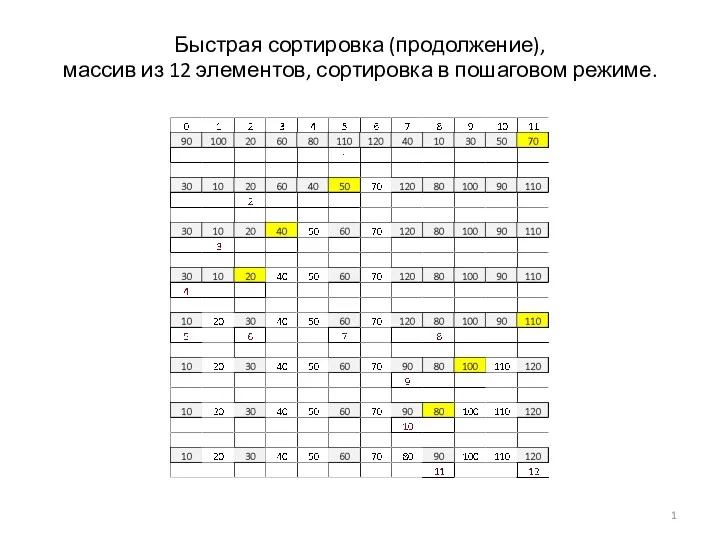 Быстрая сортировка. Массив из 12 элементов, сортировка в пошаговом режиме
Быстрая сортировка. Массив из 12 элементов, сортировка в пошаговом режиме Форматирование текста. Обработка текстовой информации. (7 класс)
Форматирование текста. Обработка текстовой информации. (7 класс) Закон Мура. Основоположники квантовой информатики
Закон Мура. Основоположники квантовой информатики Этапы создания (разработки) web-сайта
Этапы создания (разработки) web-сайта СМИ и формирование картины мира
СМИ и формирование картины мира Виды компьютерных игр
Виды компьютерных игр Команда Гугл
Команда Гугл Дистанционный урок – урок с использованием учебного материала либо полностью размещенного в Интернете
Дистанционный урок – урок с использованием учебного материала либо полностью размещенного в Интернете Текстовые документы и технологии их создания. Документ. Текстовый документ. Структурные элементы текстового документа
Текстовые документы и технологии их создания. Документ. Текстовый документ. Структурные элементы текстового документа 2 Внеклассный урок Носители информации
2 Внеклассный урок Носители информации Cистема управления версиями Git
Cистема управления версиями Git Інформатика 9 клас. Основи Інтернету
Інформатика 9 клас. Основи Інтернету Понятие как форма мышления
Понятие как форма мышления Перевод чисел в системах счисления
Перевод чисел в системах счисления Динамические данные разветвленной структуры
Динамические данные разветвленной структуры Почтовые программы
Почтовые программы Таймлайн нейронной сети
Таймлайн нейронной сети Роль СМИ в политической жизни
Роль СМИ в политической жизни Информационная безопасность. Пассивный вид атаки
Информационная безопасность. Пассивный вид атаки Онлайн – ресурсы для поиска работы и найма персонала
Онлайн – ресурсы для поиска работы и найма персонала Интеллектуальные информационные системы. Основные понятия и определения
Интеллектуальные информационные системы. Основные понятия и определения Жизненный цикл информационной системы
Жизненный цикл информационной системы Правила поведения в компьютерном классе
Правила поведения в компьютерном классе DS. Графический дизайн. Дизайн сайтов
DS. Графический дизайн. Дизайн сайтов Инструкция по работе с мобильным приложением E-Salyq Azamat в части подачи ФНО 250.00
Инструкция по работе с мобильным приложением E-Salyq Azamat в части подачи ФНО 250.00 Программное обеспечение средств защиты информации
Программное обеспечение средств защиты информации Источники поиска персонала
Источники поиска персонала