- Методы, использующиеся биоинформатикой для анализа макромолекул и создания лекарств

Содержание
- 2. Биоинформа́тика — совокупность методов и подходов[1], включающих в себя: математические методы компьютерного анализа в сравнительной геномике
- 3. Соотношение этапов развития биоинформатики
- 4. Анализ геномов – что можно извлечь из генетических текстов К настоящему времени полностью расшифрованы геномы около
- 5. Примеры организаций, которые занимаются расшифровкой геномов: The Sanger Centre, Wellcome Trust, Великобритания; The Institute of Genomic
- 6. BLAST находит области сходства между биологическими последовательностями. Программа сравнивает нуклеотидные или белковые последовательности в базы данных
- 7. В биоинформатике FASTA-формат представляет собой текстовый формат для нуклеотидных или полипептидных последовательностей, в котором нуклеотиды или
- 8. В 1999 году была опубликована первая работа, описывающая попытку выбора мишеней для действия лекарственных средств на
- 9. Программа CATS предназначена для анализа геномов с целью поиска белков, которые могли бы рассматриваться как наиболее
- 10. Три группы методов: 1. распознавание фолда (укладки, упаковки) с использованием библиотеки известных фолдов; 2. предсказания abinitio
- 11. Распознавание фолда – это стадия для построения модели трехмерной структуры белка. Оно применяется, если отсутствует информация
- 12. При предсказании abinitio целью является построение модели 3D структуры без использования знаний по структуре гомологов. Эти
- 13. В настоящее время разработано достаточно большое число различных подходов к сравнительному моделированию. Одним из наиболее широко
- 14. Сравнительная оценка различных подходов к предсказанию пространственной структуры белка по аминокислотной последовательности традиционно проводится в Асиломаре
- 15. Анализ консенсусной последовательности цитохромов Р450 семейства CYP51
- 16. Характеристика выровненных 827 последовательностей оболочечных белков E1 и E2 ВГС человека. Процентное содержание преобладающих остатков в
- 17. Методы биоинформатики эффективно используются для выяснения механизма взаимодействия макромолекул (узнавания). Методы "стыковки" (докинга) или нахождения в
- 18. Биоинформатика является базовой дисциплиной, прежде всего, при поиске мишеней действия новых лекарственных препаратов. В оценке перспективности
- 19. Моделирование взаимодействия лиганд-мишень
- 20. Комплекс нейраминидазы вируса гриппа с известным ингибитором N-ацетил-2,3-дегидро-2-деоксинейраминовой кислотой (А) и найденным путем скрининга баз данных
- 22. Скачать презентацию
![Биоинформа́тика — совокупность методов и подходов[1], включающих в себя: математические](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/111459/slide-1.jpg)
Биоинформа́тика — совокупность методов и подходов[1],
включающих в себя:
математические методы
Биоинформа́тика — совокупность методов и подходов[1],
включающих в себя:
математические методы
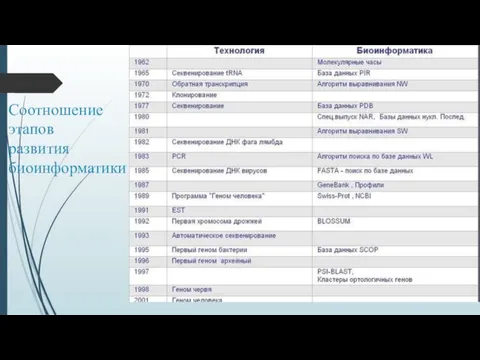
Соотношение
этапов
развития
биоинформатики
Соотношение
этапов
развития
биоинформатики

Анализ геномов – что можно извлечь из генетических текстов
К настоящему времени
Анализ геномов – что можно извлечь из генетических текстов
К настоящему времени

Примеры организаций, которые занимаются расшифровкой геномов:
The Sanger Centre, Wellcome Trust, Великобритания;
Примеры организаций, которые занимаются расшифровкой геномов:
The Sanger Centre, Wellcome Trust, Великобритания;
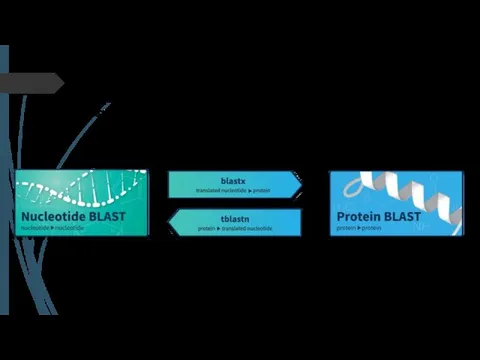
BLAST находит области сходства между биологическими последовательностями. Программа сравнивает нуклеотидные или белковые последовательности
BLAST находит области сходства между биологическими последовательностями. Программа сравнивает нуклеотидные или белковые последовательности

В биоинформатике FASTA-формат представляет собой текстовый формат для нуклеотидных или полипептидных последовательностей, в котором нуклеотиды или аминокислоты обозначаются при помощи однобуквенных кодов. Данный формат
В биоинформатике FASTA-формат представляет собой текстовый формат для нуклеотидных или полипептидных последовательностей, в котором нуклеотиды или аминокислоты обозначаются при помощи однобуквенных кодов. Данный формат

В 1999 году была опубликована первая работа, описывающая попытку выбора мишеней
В 1999 году была опубликована первая работа, описывающая попытку выбора мишеней

Программа CATS предназначена для анализа геномов с целью поиска белков, которые
Программа CATS предназначена для анализа геномов с целью поиска белков, которые

Три группы методов:
1. распознавание фолда (укладки, упаковки) с использованием библиотеки известных
Три группы методов:
1. распознавание фолда (укладки, упаковки) с использованием библиотеки известных

Распознавание фолда – это стадия для построения модели трехмерной структуры белка.
Распознавание фолда – это стадия для построения модели трехмерной структуры белка.

При предсказании abinitio целью является построение модели 3D структуры без использования
При предсказании abinitio целью является построение модели 3D структуры без использования

В настоящее время разработано достаточно большое число различных подходов к сравнительному
В настоящее время разработано достаточно большое число различных подходов к сравнительному

Сравнительная оценка различных подходов к предсказанию пространственной структуры белка по аминокислотной
Сравнительная оценка различных подходов к предсказанию пространственной структуры белка по аминокислотной
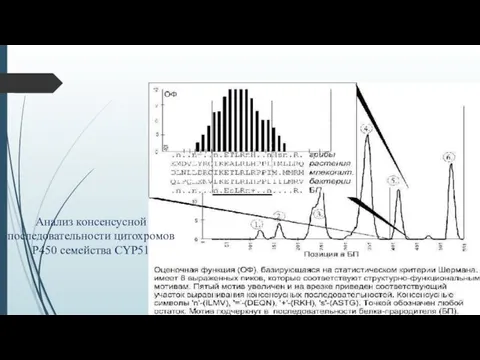
Анализ консенсусной последовательности цитохромов Р450 семейства CYP51
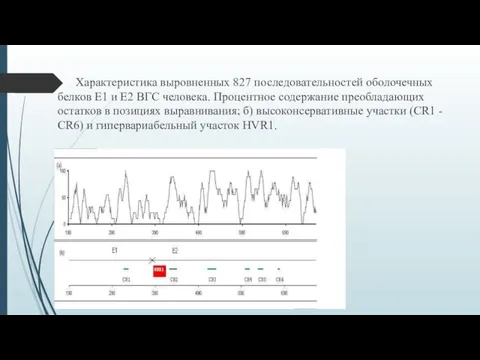
Характеристика выровненных 827 последовательностей оболочечных белков E1 и E2 ВГС человека.
Характеристика выровненных 827 последовательностей оболочечных белков E1 и E2 ВГС человека.

Методы биоинформатики эффективно используются для выяснения механизма взаимодействия макромолекул (узнавания).
Методы
Методы биоинформатики эффективно используются для выяснения механизма взаимодействия макромолекул (узнавания).
Методы

Биоинформатика является базовой дисциплиной, прежде всего, при поиске мишеней действия новых
Биоинформатика является базовой дисциплиной, прежде всего, при поиске мишеней действия новых
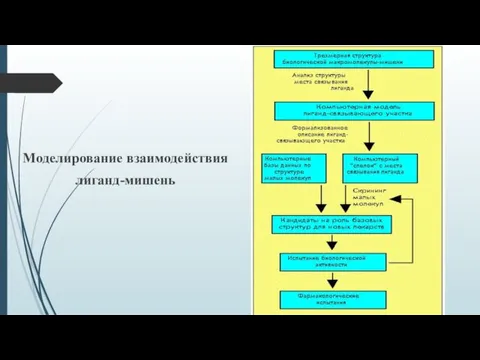
Моделирование взаимодействия
лиганд-мишень
Моделирование взаимодействия
лиганд-мишень
Комплекс нейраминидазы вируса гриппа с известным ингибитором N-ацетил-2,3-дегидро-2-деоксинейраминовой кислотой (А) и
Комплекс нейраминидазы вируса гриппа с известным ингибитором N-ацетил-2,3-дегидро-2-деоксинейраминовой кислотой (А) и
 Моделирование и представление структуры предприятия
Моделирование и представление структуры предприятия Файлы и файловые структуры
Файлы и файловые структуры Операционные системы
Операционные системы Как установить драйвер USB PopcornTV ver.2
Как установить драйвер USB PopcornTV ver.2 Разработка урока информатики в 8 кл. Текстовый редактор
Разработка урока информатики в 8 кл. Текстовый редактор Программы и программное обеспечение
Программы и программное обеспечение Програмиране за NET Framework. Отражение на типовете (Reflection)
Програмиране за NET Framework. Отражение на типовете (Reflection) Интерфейсы передачи данных
Интерфейсы передачи данных Мультиагентная технология (МАТ)
Мультиагентная технология (МАТ) Информационная безопасность в сети интернет
Информационная безопасность в сети интернет Электронная почта.
Электронная почта. Кодирование и обработка информации
Кодирование и обработка информации Основные понятия информационной безопасности и защиты информации
Основные понятия информационной безопасности и защиты информации Игра Fantasy Tournament
Игра Fantasy Tournament Визуализация информации в текстовых документах. 7 класс
Визуализация информации в текстовых документах. 7 класс SWIFT Professional Services I Alliance Lite2 Kick-off
SWIFT Professional Services I Alliance Lite2 Kick-off Прикладное программное обеспечение. Электронные таблицы MS Excel 2007
Прикладное программное обеспечение. Электронные таблицы MS Excel 2007 Путеводитель по приложению 3. Правила и процедуры работы в СМЭВ по методическим рекомендациям версии 3.х
Путеводитель по приложению 3. Правила и процедуры работы в СМЭВ по методическим рекомендациям версии 3.х NAVY. Проблемы в создании рекламных роликов
NAVY. Проблемы в создании рекламных роликов Планирование задач проекта в Microsoft Office Project
Планирование задач проекта в Microsoft Office Project Введение в CSS. Определение селекторов. Оформление текста
Введение в CSS. Определение селекторов. Оформление текста Распределённые базы данных
Распределённые базы данных Модели жизненного цикла, Каноническое и типовое проектирование ИС
Модели жизненного цикла, Каноническое и типовое проектирование ИС Программирование (Python) (§17-22). 8 класс
Программирование (Python) (§17-22). 8 класс Информационные ресурсы общества
Информационные ресурсы общества Разработка информационной системы для учета продаж билетов в авиакассах
Разработка информационной системы для учета продаж билетов в авиакассах Електронне листування з використанням веб-інтерфейсу. Вкладені файли
Електронне листування з використанням веб-інтерфейсу. Вкладені файли Растровая и векторная графика
Растровая и векторная графика