- Болезнь Шарко-Мари-Тута

Содержание
- 2. Общие данные Болезнь Шарко-Мари-Тута - обширная группа генетически гетерогенных заболеваний периферических нервов, характеризующаяся симптомами прогрессирующей полинейропатии
- 3. Причины возникновения Для наследственной нейропатии Шарко-Мари-Тута 1 типа, которая является наиболее распространенным клиническим вариантом этого заболевания,
- 4. Причины возникновения Наиболее распространенной причиной возникновения болезни (70-80% случаев) является дублирование большого региона в хромосоме 17p12,
- 5. Причины возникновения Увеличение экспрессии белка РМР22 приводит к нарушению миелинизации нервных волокон. Шванновские клетки не образуют
- 6. Патогенез Выделяют следующие виды болезни: первичная демиелинизирующая нейропатия (ШМТ1, ШМТ3, и ШМТ4) - развивается вследствие сегментарной
- 8. Симптомы I тип: начало в среднем детском возрасте; постепенно нарастает слабость в пораженных мышцах; становится заметным,
- 9. Симптомы сухожильные рефлексы снижаются и исчезают; утолщённые нервы иногда доступны пальпации; затрудняется ходьба: стоять и ходить
- 10. Симптомы иногда появляется неконтролируемая дрожь в пальцах кистей (постуральный или постурально-кинетический тремор); возможно искривление позвоночника (сколиоз
- 11. Симптомы II тип: Начало мышечной слабости в возрасте 16-30 лет Заболевание прогрессирует медленнее, чем болезнь Шарко-Мари-Тута
- 12. Наследование Тип наследования заболевания - аутосомно-доминантный, реже - аутосомно-рецессивиный, а также сцепленный с X-хромосомой. При аутосомно-доминантном
- 13. Наследование При аутосомно-рецессивном типе наследования поражаются одинаково оба пола если больны оба родителя, все дети будут
- 14. Наследование Х-сцепленный доминантный тип наследования: больные женщины передают болезнь одинаково и мальчикам, и девочкам больной мужчина
- 15. Диагностика Возраст начала заболевания, его типичная клиника, симметричный характер поражения, медленное неуклонное распространение атрофий и усугубляющаяся
- 16. Диагностика Для дифференциации ШМТ от других нервно-мышечных заболеваний (миотонии, миопатии, БАС, невропатией) проводится: электромиография (определяется снижение
- 17. Диагностика С целью исключения метаболической невропатии проводится определение сахара крови, исследование гормонов щитовидной железы, тест на
- 18. Лечение Применяется симптоматическая терапия. Проводятся повторные курсы внутримышечного введения витаминов группы В и витамина Е. С
- 19. Профилактика Профилактика наследственных амиотрофий заключается в медико-генетическом консультировании. В качестве профилактики развития ранней деформации стоп необходимо
- 21. Скачать презентацию

Общие данные
Болезнь Шарко-Мари-Тута - обширная группа генетически гетерогенных заболеваний периферических нервов,
Общие данные
Болезнь Шарко-Мари-Тута - обширная группа генетически гетерогенных заболеваний периферических нервов,

Причины возникновения
Для наследственной нейропатии Шарко-Мари-Тута 1 типа, которая является наиболее
Причины возникновения
Для наследственной нейропатии Шарко-Мари-Тута 1 типа, которая является наиболее

Причины возникновения
Наиболее распространенной причиной возникновения болезни (70-80% случаев) является
Причины возникновения
Наиболее распространенной причиной возникновения болезни (70-80% случаев) является
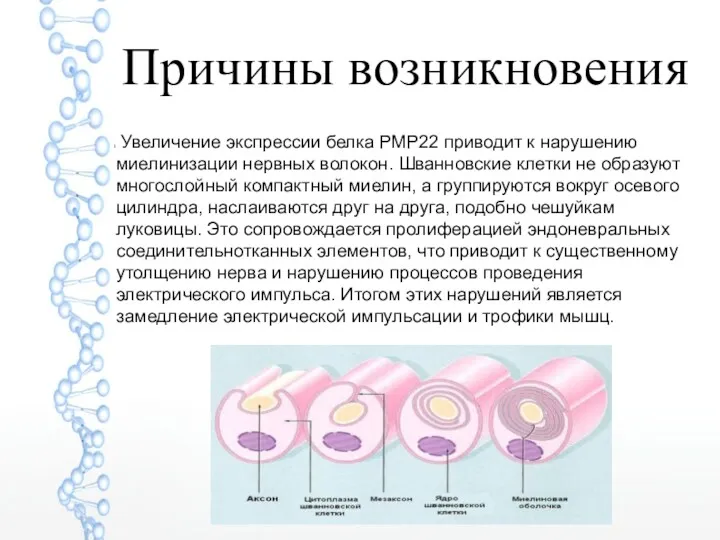
Причины возникновения
Увеличение экспрессии белка РМР22 приводит к нарушению миелинизации
Причины возникновения
Увеличение экспрессии белка РМР22 приводит к нарушению миелинизации

Патогенез
Выделяют следующие виды болезни:
первичная демиелинизирующая нейропатия (ШМТ1, ШМТ3, и ШМТ4)
Патогенез
Выделяют следующие виды болезни:
первичная демиелинизирующая нейропатия (ШМТ1, ШМТ3, и ШМТ4)


Симптомы
I тип:
начало в среднем детском возрасте;
постепенно нарастает слабость в пораженных
Симптомы
I тип:
начало в среднем детском возрасте;
постепенно нарастает слабость в пораженных

Симптомы
сухожильные рефлексы снижаются и исчезают;
утолщённые нервы иногда доступны пальпации;
затрудняется ходьба: стоять
Симптомы
сухожильные рефлексы снижаются и исчезают;
утолщённые нервы иногда доступны пальпации;
затрудняется ходьба: стоять
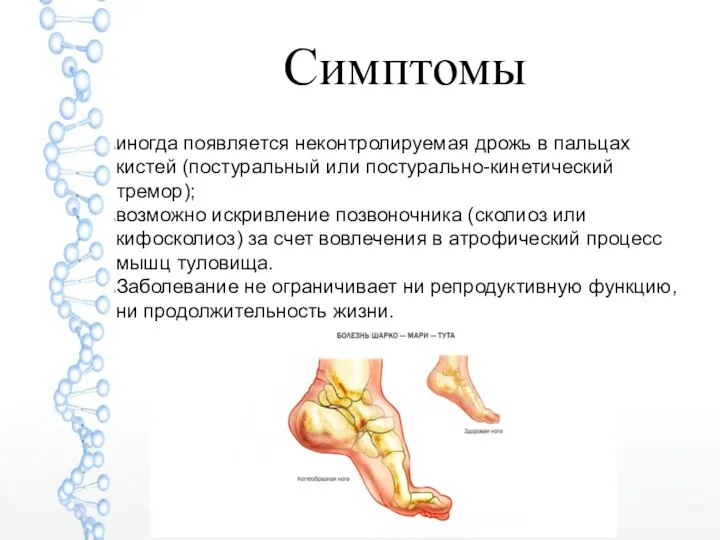
Симптомы
иногда появляется неконтролируемая дрожь в пальцах кистей (постуральный или постурально-кинетический тремор);
возможно
Симптомы
иногда появляется неконтролируемая дрожь в пальцах кистей (постуральный или постурально-кинетический тремор);
возможно
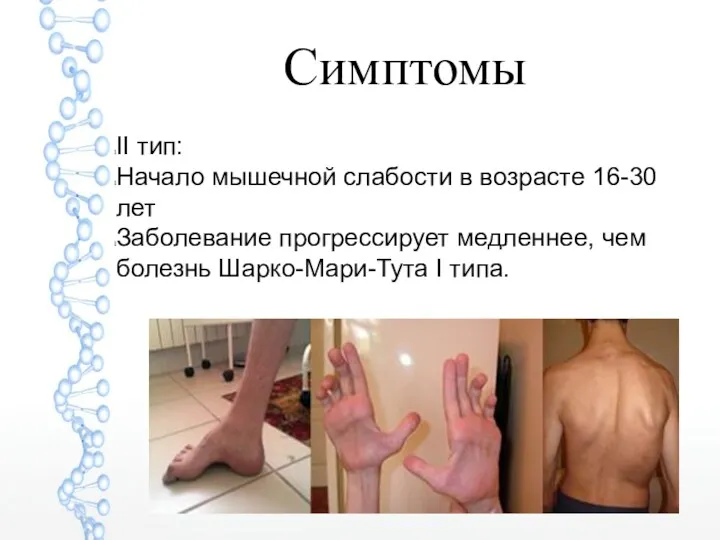
Симптомы
II тип:
Начало мышечной слабости в возрасте 16-30 лет
Заболевание прогрессирует медленнее,
Симптомы
II тип:
Начало мышечной слабости в возрасте 16-30 лет
Заболевание прогрессирует медленнее,
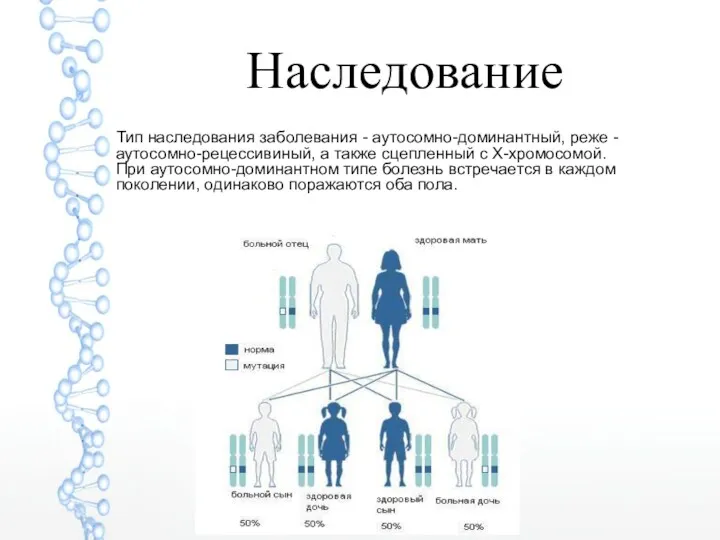
Наследование
Тип наследования заболевания - аутосомно-доминантный, реже - аутосомно-рецессивиный, а также сцепленный
Наследование
Тип наследования заболевания - аутосомно-доминантный, реже - аутосомно-рецессивиный, а также сцепленный

Наследование
При аутосомно-рецессивном типе наследования поражаются одинаково оба пола
если больны оба родителя,
Наследование
При аутосомно-рецессивном типе наследования поражаются одинаково оба пола
если больны оба родителя,

Наследование
Х-сцепленный доминантный тип наследования:
больные женщины передают болезнь одинаково и мальчикам, и
Наследование
Х-сцепленный доминантный тип наследования:
больные женщины передают болезнь одинаково и мальчикам, и

Диагностика
Возраст начала заболевания, его типичная клиника, симметричный характер поражения, медленное неуклонное
Диагностика
Возраст начала заболевания, его типичная клиника, симметричный характер поражения, медленное неуклонное

Диагностика
Для дифференциации ШМТ от других нервно-мышечных заболеваний (миотонии, миопатии, БАС, невропатией)
Диагностика
Для дифференциации ШМТ от других нервно-мышечных заболеваний (миотонии, миопатии, БАС, невропатией)

Диагностика
С целью исключения метаболической невропатии проводится определение сахара крови, исследование гормонов
Диагностика
С целью исключения метаболической невропатии проводится определение сахара крови, исследование гормонов

Лечение
Применяется симптоматическая терапия. Проводятся повторные курсы внутримышечного введения витаминов группы В
Лечение
Применяется симптоматическая терапия. Проводятся повторные курсы внутримышечного введения витаминов группы В

Профилактика
Профилактика наследственных амиотрофий заключается в медико-генетическом консультировании.
В качестве профилактики развития ранней
Профилактика
Профилактика наследственных амиотрофий заключается в медико-генетическом консультировании.
В качестве профилактики развития ранней
 Кровотечения
Кровотечения Профилактика пищевых отравлений
Профилактика пищевых отравлений Рассеянный склероз
Рассеянный склероз Опухоли. Определение понятия. Номенклатура. Биология опухолевого роста
Опухоли. Определение понятия. Номенклатура. Биология опухолевого роста Лечение когнитивных нарушений без деменции у больных дисциркуляторной энцефалопатией аксамоном
Лечение когнитивных нарушений без деменции у больных дисциркуляторной энцефалопатией аксамоном Лимфома Ходжкина. Стратификация риска и лечение
Лимфома Ходжкина. Стратификация риска и лечение Қалқанша безі гормондарының. Препараттары және антитиреоидты дәрілер
Қалқанша безі гормондарының. Препараттары және антитиреоидты дәрілер Прикладное значение биопсии
Прикладное значение биопсии Острая почечная недостаточность у детей
Острая почечная недостаточность у детей Сердечно-сосудистые заболевания. Тема 5
Сердечно-сосудистые заболевания. Тема 5 Өндірістік токсикология негіздері. (Тақырып 8)
Өндірістік токсикология негіздері. (Тақырып 8) Кровь. Лимфа. Кроветворение (гемопоэз)
Кровь. Лимфа. Кроветворение (гемопоэз) СНІД, або сидром набутого імунодефіциту
СНІД, або сидром набутого імунодефіциту Иммобилизация және зардап шеккен адамды тасымалдау
Иммобилизация және зардап шеккен адамды тасымалдау СТЖБ-ң анатомиясы, физиологиясы; СТЖБ-ң қызметінің бұзылысы; Артикулатемпералды синдром
СТЖБ-ң анатомиясы, физиологиясы; СТЖБ-ң қызметінің бұзылысы; Артикулатемпералды синдром ЛФК при заболеваниях сердечно-сосудистой системы
ЛФК при заболеваниях сердечно-сосудистой системы Психбелсенді заттардың жіктелуі. Нашақорлық синдромы
Психбелсенді заттардың жіктелуі. Нашақорлық синдромы Медична гельмінтологія. Круглі та плоскі черви –паразити людини. Лекція 9
Медична гельмінтологія. Круглі та плоскі черви –паразити людини. Лекція 9 Қазіргі жаратылыстану концепциялары пәні және әлеуметтік маңызы. Ғылыми дүниетанудың ерекшеліктері мен құрылысы
Қазіргі жаратылыстану концепциялары пәні және әлеуметтік маңызы. Ғылыми дүниетанудың ерекшеліктері мен құрылысы Анемии
Анемии Принципы купирования острого инфаркта миокарда
Принципы купирования острого инфаркта миокарда Вспомогательные репродуктивные технологии
Вспомогательные репродуктивные технологии Средства, влияющие на эфферентную иннервацию. Адренергические средства
Средства, влияющие на эфферентную иннервацию. Адренергические средства Алкоголизм. Этиология и патогенез
Алкоголизм. Этиология и патогенез Чума. Возбудитель чумы
Чума. Возбудитель чумы Профессиональные заболевания с преимущественным поражением почек и мочевыводящих путей
Профессиональные заболевания с преимущественным поражением почек и мочевыводящих путей Закаливание детей в ДОУ
Закаливание детей в ДОУ Пластическая хирургия
Пластическая хирургия