- Диагностика и лечение эпилептических энцефалопатий детского возраста

Содержание
- 2. Ранняя младенческая эпилептическая энцефалопатия (синдром Отахара) Этиология: пренатальный поражение ГМ (врожденные мальформации, нейро-кожные синдромы, подострая диффузная
- 3. Диагностика синдрома Отахара При неврологическом осмотре: задержка психического и моторного развития, геми- или тетрапарез, атрофия зрительного
- 4. Лечение синдрома Отахара Стартовая терапия – топирамат. Топамакс в высоких дозах (10-15 мг/кг/сут) в 2 приёма.
- 5. Синдром Отахара Возможны комбинации топирамата с вальпроатами или барбитуратами, бензодиазепинами. При полной неэффективности АЭП возможна их
- 6. Ранняя миоклоническая энцефалопатия (синдром Айкарди) Этиология: определенную роль могут играть врожденные нарушения метаболизма Дебют: в первые
- 7. Диагностика синдрома Айкарди В неврологическом статусе: задержка психомоторного развития, диффузная мышечная гипотония. ЭЭГ исследование: паттерн вспышка-угнетение
- 8. Лечение синдрома Айкарди Стартовая терапия – вальпроаты. Конвулекс в дозе 30-100мг/кг/сут в 3 приёма. Препараты второго
- 9. Синдром Веста Симптоматическая, или предположительно, симптоматическая, генерализованная форма эпилепсии. Этиология: мальформация сосудов ГМ изолированная или в
- 10. Диагностические критерии синдрома Веста Особый тип эпилептических приступов – инфантильные спазмы (массивные миоклонические и (или) тонические,
- 11. Диагностика синдрома Веста Неврологический статус: задержка психического и моторного развития, центральные парезы и параличи, косоглазие, микроцефалия
- 12. Дифференциальная диагностика
- 13. Лечение синдрома Веста Стартовое лечение – вигабатрин. Сабрил в дозе 50-150 мг/кг/сут в 2 приема. Препарат
- 14. Тяжелая миоклоническая эпилепсия младенчества (синдром Драве) Криптогенный эпилептический синдром, имеющий черты как генерализованные, так и фокальные
- 15. Диагностика синдрома Драве Неврологический статус: мышечная гипотония, атаксия, признаки пирамидной недостаточности, фотосенситивность, задержка психического и речевого
- 16. Лечение синдрома Драве Стартовое лечение – топирамат. Сабрил в дозе 3-10 мг/кг/сут в 2 приема. Препарат
- 18. Синдром Ландау-Клеффнера (приобретенная эпилептическая афазия) Предположительно идиопатическая форма эпилепсии Дебют: 3-7 лет. Речевые нарушения – кардинальный
- 19. Диагностика синдрома Ландау-Клеффнера Неврологический статус: сенсорная или тотальная афазия, нарушение поведения. ЭЭГ: высокоамплитудные региональные острые волны
- 20. Дифференциальная диагностика
- 21. Лечение синдрома Ландау-Клеффнера При эпилептической афазии без эпилептических приступов: монотерапия сукцинимидами или бензодиазепинами При сочетании с
- 23. Синдром Леннокса-Гасто Генерализованная криптогенная или симптоматическая форма эпилепсии. Дебют: от 2 до 8 лет, пик –
- 24. Диагностика синдрома Леннокса-Гасто Неврологический статус: при криптогенном варианте очаговые симптомы отсутствуют, при симптоматическом могут быть центральные
- 25. Лечение синдрома Леннокса-Гасто Стартовая терапия: топирамат. Топамакс 3-10 мг/кг/сут в 2 приема Препарат второго выбора: вальпроевая
- 26. Синдром псевдо-Леннокса Предположительно идиопатическая форма эпилепсии В 5% роландическая эпидлепсия может трансформироваться в СПЛ. Дебют: от
- 27. Диагностика синдрома псевдо-Леннокса Неврологическое обследование: динамическая атаксия, атаксия, дисметрия, брадикинезия, когнитивные и речевые расстройства. ЭЭГ: высокоамплитудная
- 28. Лечение синдрома псевдо-Леннокса Стартовое лечение: вальпроевая кислота (Конвульсофин в дозе 30-70 мг/кг/сут в 2-3 приема. Препараты
- 31. Скачать презентацию

Ранняя младенческая эпилептическая энцефалопатия (синдром Отахара)
Этиология: пренатальный поражение ГМ (врожденные мальформации,
Ранняя младенческая эпилептическая энцефалопатия (синдром Отахара)
Этиология: пренатальный поражение ГМ (врожденные мальформации,

Диагностика синдрома Отахара
При неврологическом осмотре: задержка психического и моторного развития, геми-
Диагностика синдрома Отахара
При неврологическом осмотре: задержка психического и моторного развития, геми-

Лечение синдрома Отахара
Стартовая терапия – топирамат. Топамакс в высоких дозах (10-15
Лечение синдрома Отахара
Стартовая терапия – топирамат. Топамакс в высоких дозах (10-15

Синдром Отахара
Возможны комбинации топирамата с вальпроатами или барбитуратами, бензодиазепинами.
При полной
Синдром Отахара
Возможны комбинации топирамата с вальпроатами или барбитуратами, бензодиазепинами.
При полной

Ранняя миоклоническая
энцефалопатия (синдром Айкарди)
Этиология: определенную роль могут играть врожденные нарушения
Ранняя миоклоническая
энцефалопатия (синдром Айкарди)
Этиология: определенную роль могут играть врожденные нарушения

Диагностика синдрома Айкарди
В неврологическом статусе: задержка психомоторного развития, диффузная мышечная гипотония.
ЭЭГ
Диагностика синдрома Айкарди
В неврологическом статусе: задержка психомоторного развития, диффузная мышечная гипотония.
ЭЭГ

Лечение синдрома Айкарди
Стартовая терапия – вальпроаты. Конвулекс в дозе 30-100мг/кг/сут в
Лечение синдрома Айкарди
Стартовая терапия – вальпроаты. Конвулекс в дозе 30-100мг/кг/сут в

Синдром Веста
Симптоматическая, или предположительно, симптоматическая, генерализованная форма эпилепсии.
Этиология: мальформация сосудов ГМ
Синдром Веста
Симптоматическая, или предположительно, симптоматическая, генерализованная форма эпилепсии.
Этиология: мальформация сосудов ГМ

Диагностические критерии синдрома Веста
Особый тип эпилептических приступов – инфантильные спазмы
Диагностические критерии синдрома Веста
Особый тип эпилептических приступов – инфантильные спазмы

Диагностика синдрома Веста
Неврологический статус: задержка психического и моторного развития, центральные парезы
Диагностика синдрома Веста
Неврологический статус: задержка психического и моторного развития, центральные парезы

Дифференциальная диагностика
Дифференциальная диагностика

Лечение синдрома Веста
Стартовое лечение – вигабатрин. Сабрил в дозе 50-150 мг/кг/сут
Лечение синдрома Веста
Стартовое лечение – вигабатрин. Сабрил в дозе 50-150 мг/кг/сут

Тяжелая миоклоническая эпилепсия младенчества (синдром Драве)
Криптогенный эпилептический синдром, имеющий черты как
Тяжелая миоклоническая эпилепсия младенчества (синдром Драве)
Криптогенный эпилептический синдром, имеющий черты как

Диагностика синдрома Драве
Неврологический статус: мышечная гипотония, атаксия, признаки пирамидной недостаточности, фотосенситивность,
Диагностика синдрома Драве
Неврологический статус: мышечная гипотония, атаксия, признаки пирамидной недостаточности, фотосенситивность,

Лечение синдрома Драве
Стартовое лечение – топирамат. Сабрил в дозе 3-10 мг/кг/сут
Лечение синдрома Драве
Стартовое лечение – топирамат. Сабрил в дозе 3-10 мг/кг/сут
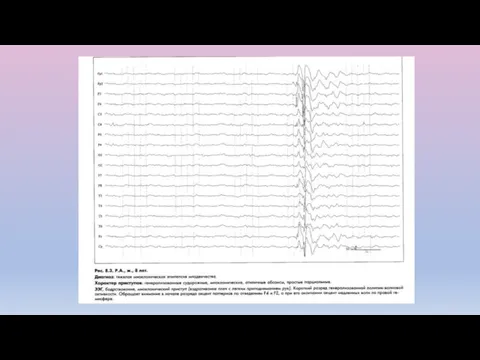

Синдром Ландау-Клеффнера (приобретенная эпилептическая афазия)
Предположительно идиопатическая форма эпилепсии
Дебют: 3-7 лет.
Речевые нарушения
Синдром Ландау-Клеффнера (приобретенная эпилептическая афазия)
Предположительно идиопатическая форма эпилепсии
Дебют: 3-7 лет.
Речевые нарушения

Диагностика синдрома Ландау-Клеффнера
Неврологический статус: сенсорная или тотальная афазия, нарушение поведения.
ЭЭГ: высокоамплитудные
Диагностика синдрома Ландау-Клеффнера
Неврологический статус: сенсорная или тотальная афазия, нарушение поведения.
ЭЭГ: высокоамплитудные

Дифференциальная диагностика
Дифференциальная диагностика

Лечение синдрома Ландау-Клеффнера
При эпилептической афазии без эпилептических приступов: монотерапия сукцинимидами или
Лечение синдрома Ландау-Клеффнера
При эпилептической афазии без эпилептических приступов: монотерапия сукцинимидами или
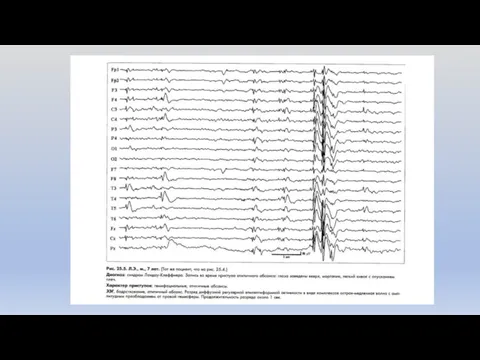

Синдром Леннокса-Гасто
Генерализованная криптогенная или симптоматическая форма эпилепсии.
Дебют: от 2 до 8
Синдром Леннокса-Гасто
Генерализованная криптогенная или симптоматическая форма эпилепсии.
Дебют: от 2 до 8

Диагностика синдрома Леннокса-Гасто
Неврологический статус: при криптогенном варианте очаговые симптомы отсутствуют, при
Диагностика синдрома Леннокса-Гасто
Неврологический статус: при криптогенном варианте очаговые симптомы отсутствуют, при

Лечение синдрома Леннокса-Гасто
Стартовая терапия: топирамат. Топамакс 3-10 мг/кг/сут в 2 приема
Препарат
Лечение синдрома Леннокса-Гасто
Стартовая терапия: топирамат. Топамакс 3-10 мг/кг/сут в 2 приема
Препарат

Синдром псевдо-Леннокса
Предположительно идиопатическая форма эпилепсии
В 5% роландическая эпидлепсия может трансформироваться в
Синдром псевдо-Леннокса
Предположительно идиопатическая форма эпилепсии
В 5% роландическая эпидлепсия может трансформироваться в

Диагностика синдрома псевдо-Леннокса
Неврологическое обследование: динамическая атаксия, атаксия, дисметрия, брадикинезия, когнитивные и
Диагностика синдрома псевдо-Леннокса
Неврологическое обследование: динамическая атаксия, атаксия, дисметрия, брадикинезия, когнитивные и

Лечение синдрома псевдо-Леннокса
Стартовое лечение: вальпроевая кислота (Конвульсофин в дозе 30-70 мг/кг/сут
Лечение синдрома псевдо-Леннокса
Стартовое лечение: вальпроевая кислота (Конвульсофин в дозе 30-70 мг/кг/сут
 Показания к переводу на искусственную вентиляцию легких. Параметры КЩС. Режимы вентиляции
Показания к переводу на искусственную вентиляцию легких. Параметры КЩС. Режимы вентиляции Дәрігелік құпия және дәрігерлік қателік
Дәрігелік құпия және дәрігерлік қателік Эндокриндік патологияның визуалді диагностика әдістерінің ерекшеліктері
Эндокриндік патологияның визуалді диагностика әдістерінің ерекшеліктері Медицинская этика, понятия, принципы, значимость в медицинской практики. Этические аспекты взаимоотношений врача и пациента
Медицинская этика, понятия, принципы, значимость в медицинской практики. Этические аспекты взаимоотношений врача и пациента Polycythemia
Polycythemia Черви-паразиты. Тест
Черви-паразиты. Тест Повреждения: некроз, атрофия, апоптоз
Повреждения: некроз, атрофия, апоптоз Гангрена, некроз, язвы, свищи
Гангрена, некроз, язвы, свищи Шовные материалы
Шовные материалы Политравма. Особенности. Диагностика
Политравма. Особенности. Диагностика Основные принципы лечения локализованного и генерализованного пародонтита
Основные принципы лечения локализованного и генерализованного пародонтита Влияние музыки на здоровье человека
Влияние музыки на здоровье человека Гендік аурулар
Гендік аурулар Дерматологияда жергілікті емде қолданылатын дәрілік заттар
Дерматологияда жергілікті емде қолданылатын дәрілік заттар Экстракорпоральная мембранная оксигенация
Экстракорпоральная мембранная оксигенация Пиодермии. Определение
Пиодермии. Определение Пищевод. Желудок. Тонкая кишка. Хирургическая анатомия, лимфоотток. Оперативные доступы, принципы малоинвазивной хирургии
Пищевод. Желудок. Тонкая кишка. Хирургическая анатомия, лимфоотток. Оперативные доступы, принципы малоинвазивной хирургии Мониторинг состояния пациента
Мониторинг состояния пациента Pancreatic Cancer
Pancreatic Cancer Инновационные технологии в абилитации и реабилитации детей с нарушениями центральной и периферической нервной системы
Инновационные технологии в абилитации и реабилитации детей с нарушениями центральной и периферической нервной системы Радионуклидная диагностика головного мозга
Радионуклидная диагностика головного мозга Ожоги. Отморожения. Электротравма. Первая помощь, профилактика. (Лекция 12)
Ожоги. Отморожения. Электротравма. Первая помощь, профилактика. (Лекция 12) Аллергия. Тесты. Задачи
Аллергия. Тесты. Задачи Ишемический инсульт
Ишемический инсульт Амбулаторные операции в полости рта
Амбулаторные операции в полости рта Сестринская помощь при заболеваниях ЛОР-органов
Сестринская помощь при заболеваниях ЛОР-органов Кадровая политика в области здравоохранения
Кадровая политика в области здравоохранения Патофизиология водно-солевого обмена
Патофизиология водно-солевого обмена