- Фенилкетонурия

Содержание
- 2. Содержание: Фенилкетонурия. Признаки. Причины. Диагностика фенилкетонурии. Лечение. Список литературы.
- 3. Фенилкетонурия Наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина.
- 6. Признаки. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка
- 9. Причины. Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном
- 11. Диагностика фенилкетонурии. В настоящее время диагностика фенилкетонурии входит в программу неонатального скрининга, осуществляемого всем новорожденным. Для
- 13. Лечение. Соблюдение диеты, ограничивающей поступление белка в организм. Больным назначается прием минеральных соединений, витаминов группы В
- 18. Скачать презентацию

Содержание:
Фенилкетонурия.
Признаки.
Причины.
Диагностика фенилкетонурии.
Лечение.
Список литературы.
Содержание:
Фенилкетонурия.
Признаки.
Причины.
Диагностика фенилкетонурии.
Лечение.
Список литературы.

Фенилкетонурия
Наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме
Фенилкетонурия
Наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме
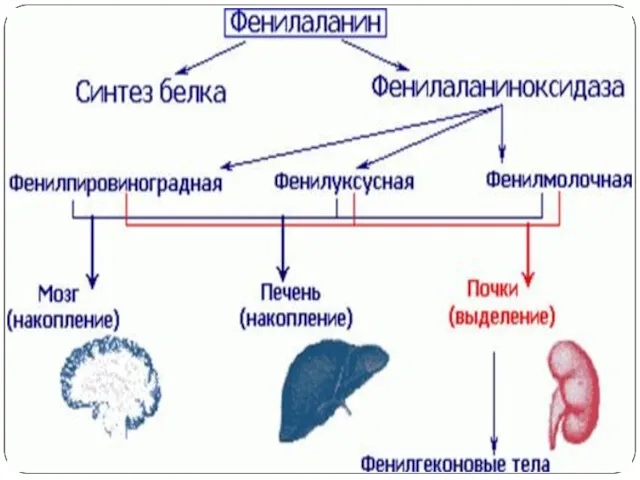

Признаки.
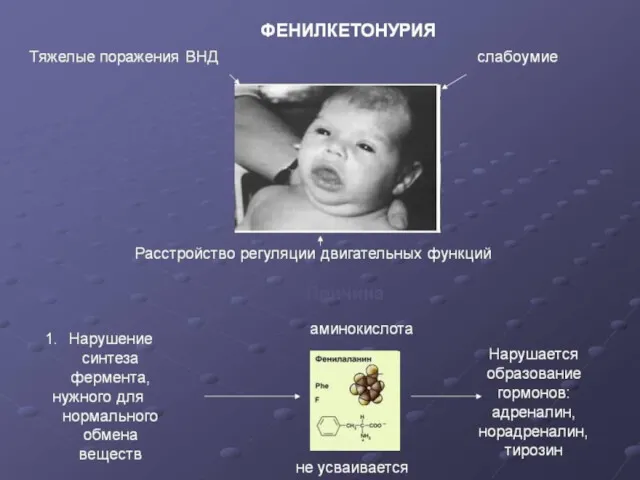
Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от
Признаки.
Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от



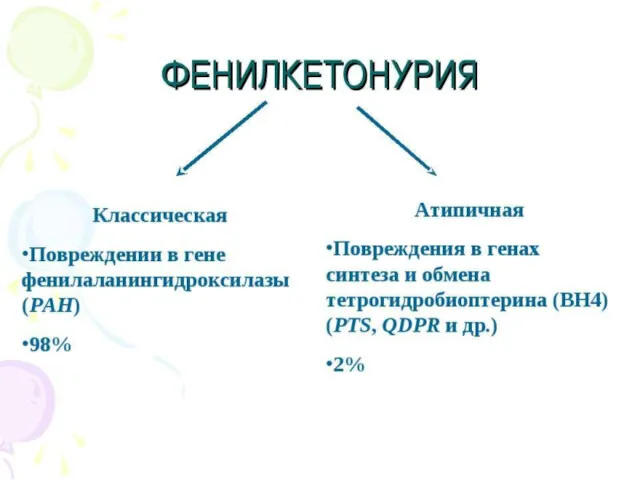
Причины.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу
Причины.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу

Диагностика фенилкетонурии.
В настоящее время диагностика фенилкетонурии входит в программу неонатального скрининга,
Диагностика фенилкетонурии.
В настоящее время диагностика фенилкетонурии входит в программу неонатального скрининга,


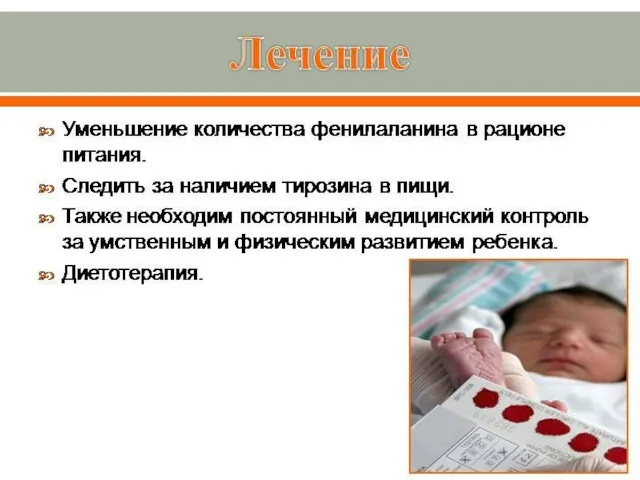
Лечение.
Соблюдение диеты, ограничивающей поступление белка в организм.
Больным назначается прием
Лечение.
Соблюдение диеты, ограничивающей поступление белка в организм.
Больным назначается прием


 Патофизиология половых желез
Патофизиология половых желез Влияние климата на здоровье людей
Влияние климата на здоровье людей 4LifeTransform Бёрн™ в программах снижения веса
4LifeTransform Бёрн™ в программах снижения веса Значение открытого овального окна в возникновении криптогенного инсульта
Значение открытого овального окна в возникновении криптогенного инсульта Особенности оказания медико-психологической и реабилитационной помощи комбатантам в Российской Федерации
Особенности оказания медико-психологической и реабилитационной помощи комбатантам в Российской Федерации Классификация грыж живота
Классификация грыж живота Тики (тикозные гиперкинезы)
Тики (тикозные гиперкинезы) Периодонтит временных зубов у детей
Периодонтит временных зубов у детей СП при атеросклерозе
СП при атеросклерозе Эндопротезирование тазобедренного сустава
Эндопротезирование тазобедренного сустава Хвороби в ділянці голови великої рогатої худоби
Хвороби в ділянці голови великої рогатої худоби Методы оценки функционального состояния. Тема 3
Методы оценки функционального состояния. Тема 3 Ферменты и гормоны, витамины, лекарства
Ферменты и гормоны, витамины, лекарства Синдром кошачьего крика (Синдром Лежена)
Синдром кошачьего крика (Синдром Лежена) Диагностика и лечение аритмий (А) и блокад (Б) сердца
Диагностика и лечение аритмий (А) и блокад (Б) сердца Тактика ведения ВИЧ-инфицированных пациентов с туберкулезом. Лечение туберкулеза у ВИЧ-инфицированных
Тактика ведения ВИЧ-инфицированных пациентов с туберкулезом. Лечение туберкулеза у ВИЧ-инфицированных Бактериальный бронхит
Бактериальный бронхит Зәр шығару жүйесі
Зәр шығару жүйесі Стафилококки. Стрептококки. Менингококки. Гонококки
Стафилококки. Стрептококки. Менингококки. Гонококки Правила профилактики гриппа и коронавирусной инфекции
Правила профилактики гриппа и коронавирусной инфекции Средства, влияющие на иммунитет
Средства, влияющие на иммунитет Сердечно-сосудистые осложнения при диффузном токсическом зобе
Сердечно-сосудистые осложнения при диффузном токсическом зобе Гинекологиялық науқастардан анамнез жинау
Гинекологиялық науқастардан анамнез жинау Радиоизотопная диагностика
Радиоизотопная диагностика Хронический гастрит, язвенная болезнь, рак желудка
Хронический гастрит, язвенная болезнь, рак желудка Медична інформаційна система
Медична інформаційна система Головокружение и нарушение устойчивости. Современные методы обследования и лечения
Головокружение и нарушение устойчивости. Современные методы обследования и лечения Специфика логопедической работы с детьми с расстройствами аутистического спектра
Специфика логопедической работы с детьми с расстройствами аутистического спектра