- Генетика и наследственные заболевания человека

Содержание
- 2. Прогресс науки и техники подвергает современных людей существенно большим рискам неблагоприятной изменчивости. Физические, химические и биологические
- 3. Многие мутации генов и практически все аберрации хромосом неблагоприятны как для индивида, так и для популяции;
- 4. К настоящему времени описано свыше 3500 наследственных заболеваний. Около 5-5,5% детей рождаются с наследственной или врожденной
- 5. МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА 1. Клинико-генеалогический метод (составление родословных, предложил в1865 г. Ф.Гальтон). 2. Близнецовый метод
- 6. Клинико-генеалогический метод - здоровый мужчина (больной - ) - здоровая женщина (больная - ) Метод состоит
- 7. Близнецовый метод Двойни встречаются 1/84 новорожденных, 1/3 из них – монозиготные (однояйцовые – близнецы), остальные -
- 8. Коэффициент интеллекта, или IQ, позволяет количественно измерить генетически обусловленные умственные способности людей ( у дизиготных близнецов
- 9. Дерматоглифический метод В генетике используются разделы: дактилоскопия (рис. на подушечках пальцев), пальмоскопия (рис. на ладонях) и
- 10. Цитогенетический метод В 1956 г. швед. ученые Д. Тийо и А Левин разработали метод культивирования человеческих
- 11. Наследственные заболевания человека
- 12. Заболевания, обусловленные изменениями числа и структуры хромосом(генные и хромосомные мутации соответственно), называются хромосомными Чем больше хромосомного
- 13. Аутосомно-доминантное наследование Этот тип наследования лежит в основе ряда заболеваний, сопровождающихся нарушением синтеза структурных белков и
- 14. Основные понятия,используемые при характеристике заболеваний Эпикант (эпикантус) - вертикальная складка кожи полулунной формы, прикрывающая внутренний угол
- 15. Акроцефалия - высокий «башенный» череп Алопеция – стойкое или временное выпадение волос Аменорея – отсутствие менструального
- 16. Прогерия – преждевременное старение организма Птеригиум – крыловидные складки кожи Птоз – опущение внутренних органов или
- 17. МИКРОСОМИЯ Синдром первой жаберной дуги. Клинические признаки: односторонняя аномалия ушной раковины и гипоплазия нижней челюсти; аномалии
- 18. СИНДРОМ РОБИНОВА Впервые описан в 1969 г. Клинические признаки: необычное строение лица, умеренная карликовость, гипоплазия половых
- 19. СИНДРОМ ВИЛЬЯМСА Впервые описан в 1961 г. Клинические признаки: Необычное лицо, низкий рост, короткий нос, полные
- 20. СИНДРОМ МАРФАНА Впервые описан в 1896 г. Клинические признаки: высокий рост, арахнодактилия, подвывих хрусталика, порок митрального
- 21. Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от аневризма аорты. Единственная компенсация –
- 22. АКРОЦЕФАЛОСИНДАКТИЛИЯ Клинические признаки: изменение черепа, гипоплазия основания черепа, плоский лоб, гипертелоризм, запавшая переносица, синдактилия, косоглазие, слабоумие.
- 23. Трихо-рино-фалангетальный синдром Клинические признаки: отставание в росте, лицо с грушевидным носом, оттопыренные уши, редкие, тонкие и
- 24. ПОЛИДАКТИЛИЯ Клинические признаки: существует два варианта: тип А, при котором дополнительный палец функционален, и тип В,
- 25. СИНДАКТИЛИЯ Клинические признаки: синдактилия – это сращение различных пальцев кистей и стоп. На кистях чаще всего
- 26. ОСТЕОГЕНЕЗ Клинические признаки: повышенная ломкость трубчатых костей, ребер и ключич при минимальной травме, деформации конечностей, голубые
- 27. МИОТОНИЧЕСКАЯ ДИСТРОФИЯ Миотоническая дистрофия, или болезнь Штейнерта – многосистемное заболевание у обоих полов. Клинические признаки: миотония,
- 28. ЭКТРОДАКТИЛИЯ Впервые описан в 1970 г. Клинические признаки: недоразвитие или отсутствие одного или нескольких пальцев кистей
- 29. СИНДРОМ КРУЗОНА (черепно-лицевой дизостоз) Синдром Крузона – дефект гена каспазы, 10q. Впервые описан в 1912 г.
- 30. АХОНДРОПЛАЗИЯ Клинические признаки: диспропорциональная карликовость (рост 120-130 см) за счет укорочения конечностей, большой череп, кисти широкие
- 31. ВИТИЛИГО Клинические признаки: частичная депигментация кожи; поражение обычно симметричное на руках, лице, шее. Больные очень чувствительны
- 32. ГИПЕРТРИХОЗ («ЛЮДИ – ВОЛКИ») Клинические признаки: чрезмерный рост волос на всех частях тела, кроме ладоней и
- 33. Порфирия, или вампиризм "..Ученые выяснили, что вампиризм – это тяжелое, очень редкое заболевание – порфирия, которая
- 34. СИНДРОМ ТРИЧЕРА КОЛЛИНЗА (челюстно-лицевой дизостоз) Аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией. Встречается у 1 из 50 000
- 35. НЕЙРОФИБРОМАТОЗ (болезнь Реклингаузена) а НФ1 характеризуется образованием опухолей на нервных тканях, и, вызывающее различные кожные и
- 36. Аутосомно-рецессивный тип наследования 1. Больной ребенок рождается у клинически здоровых родителей. 2. Болеют сибсы, т.е. братья
- 37. АЛЬБИНИЗМ Аутосомно-рецессивное заболевание, характеризующееся недостаточным содержанием пигмента меланина и влияющее на кожу и глаза. Иногда поражаются
- 38. Группа заболеваний, обусловленных наследственной патологией соединительной ткани, различающихся по характеру обменных нарушений, но имеющих большое клиническое
- 39. АХОНДРОГЕНЕЗ Клинические признаки: водянка плода, резкое укорочение конечностей, шеи и туловища, большие размеры черепа. Рентгенологически выявляется
- 40. Синдром Лоуренса-Муна-Барде-Бидля Впервые описан в 1866 г. J. Laurence и R. Moon. Клинические признаки:ожирение, гипогонадизм, умственная
- 41. РАСЩЕЛИНА ГУБЫ Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, часто эпикант и телоризм, деформации первых пальцев
- 42. СИНДРОМ АПЕРТА (череп в форме трилистника) Клинические признаки: характерная форма черепа (возникает вследствие внутриутробного зарастания швов)
- 43. СИНДРОМ НУНАН Впервые описан в 1928 г. Клинические признаки: гипертелоризм, эпикант, низко посаженные уши, нарушение прикуса,
- 44. СИНДРОМ КОККЕЙНА Впервые описан в 1946 г. Клинические признаки: низкорослость, старообразное лицо, микроцефалия, умствен - ная
- 45. КСЕРОДЕРМА ПИГМЕНТНАЯ (дерматоз Капоши) Пигментная ксеродерма – заболевание, протекающее с поражением кожи, фоточувствительностью, злокачественными новообразованиями. Клинические
- 46. МУКОПОЛИСАХАРИДОЗ Синдром Моркио описан в 1929 г. Клинические признаки: отставание в росте, деформация позвоночника и грудины,
- 47. ФЕНИЛКЕТОНУРИЯ Фенилкетонурия – болезнь аминокислотного обмена. Описана в 1934 г. А. Фелингом. Патология связана с недостаточностью
- 48. Доминантное наследование, сцепленное с X-хромосомой-этот тип наследования прослеживается, например, при фосфат-диабете. Рецессивное наследование, сцепленное с X-хромосомой-этот
- 49. Действие мутантного гена проявляется только при XY-наборе половых хромосом, т.е. у мальчиков. Вероятность рождения больного мальчика
- 50. Болезнь Фабри Является редкой наследственной Х-сцепленной рецессивной лизосомальной болезнью накопления. Болезнь поражает гемизиготных мужчин (т.е. всех
- 51. Действие доминантного мутантного гена проявляется в любом наборе половых хромосом (XX, XY, ХО и др.). Заболевание
- 52. ФОСФАТ-ДИАБЕТ Передается доминантным путем, сцепленным с Х-хромосомой. Наследственное рахитоподобное заболевание, обусловленное нарушением всасывания кальция и фосфора
- 53. ХРОМОСОМНЫЕ БОЛЕЗНИ Хромосомные заболевания связаны с аномалиями числа или структуры хромосом. Для них характерно: малый рост
- 54. РОДОСЛОВНАЯ С Х-СЦЕПЛЕННЫМ ТИПОМ НАСЛЕДОВАНИЯ 1. Болеют только мальчики по линии матери. 2. Родители пробанда здоровы.
- 55. ГИДРОЦЕФАЛИЯ Клинические признаки: увеличение объема головы, расширение желудочков мозга; истончение и расхождение костей черепа, диспропорция мозговой
- 56. ГЕМОФИЛИЯ А Клинические признаки:под- и внутри кожные кровотечения, кровоизлияния в крупные суставы, подкожные и межмышечные гематомы,
- 57. СИНДРОМ ДАУНА (ТРИСОМИЯ 21) Описан в 1866 г. Клинические признаки: умственная отсталость, плоское лицо, монголоид ный
- 58. СИНДРОМ МАРТИНА-БЕЛЛА Синдром Мартина-Белла – самая распространенная (после болезни Дауна) форма умственной отсталости. Мальчики болеют в
- 59. СИНДРОМ ААРСКОГО Синдром Аарского, или лице-пальце-генитальный синдром подобно описан в 1970 г. Клинические признаки: отставание в
- 60. СИНДРОМ КОФФИНА-ЛОУРИ Синдром впервые описан в 1966 г. Клинические признаки: антимонголоидный разрез глаз, гипертелоризм, луковицеобразный нос,
- 61. Синдром трисомии 9р Клинические признаки: умственная отсталость, задержка роста, микробрахице-фалия, антимонголоидный разрез глаз, глубоко посаженные глаза
- 62. СИНДРОМ КЛАЙНФЕЛЬТЕРА (47, ХХУ) Описан в 1942 г. Клинические признаки: высокий рост, хрупкое телосложение, гипоплазия яичек,
- 63. СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА (ХО –СИНДРОМ) Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые вторичные половые признаки, крыловидные
- 64. СИНДРОМ ПАТАУ (ТРИСОМИЯ 13) Описан в 1961 г. Клинические признаки: микроцефалия, расщепле –ние губы и неба,
- 65. Синдром Эдвардса – трисомия 18 Клинические признаки: задержка пренатального развития, множественные пороки развития черепа (маленькая нижняя
- 66. СИНДРОМ ЛЕЖЕНА (кошачьего крика,моносомия 5р) Описан в 1963 г. Клинические признаки: необычный плач, напоминающий кошачье мяуканье,
- 67. СИНДРОМ СВАЕРА (ДИСГЕНЕЗИЯ ГОНАД, ХУ ТИП ) Клинические признаки: наружные половые органы сформированы по женскому типу,
- 68. СИНДРОМ ЭЛЕРСА-ДАНЛОСА Описан в 1657 г. Клинические признаки: гиперрастяжимость соединительной ткани (нарушение синтеза коллагена); кожа тонкая
- 69. ПРОГЕРИЯ Описана в 1886 г. Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения в 8-10 раз.
- 70. Основным способом предотвращения генетических заболеваний является тщательное клиническое обследование молодожёнов (в семьях которых есть генетически неблагополучные
- 72. Скачать презентацию

Прогресс науки и техники подвергает современных людей существенно большим рискам неблагоприятной
Прогресс науки и техники подвергает современных людей существенно большим рискам неблагоприятной

Многие мутации генов и практически все аберрации хромосом неблагоприятны как для
Многие мутации генов и практически все аберрации хромосом неблагоприятны как для

К настоящему времени описано свыше 3500 наследственных заболеваний. Около 5-5,5% детей
К настоящему времени описано свыше 3500 наследственных заболеваний. Около 5-5,5% детей

МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
1. Клинико-генеалогический метод (составление родословных, предложил в1865 г.
МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ ЧЕЛОВЕКА
1. Клинико-генеалогический метод (составление родословных, предложил в1865 г.
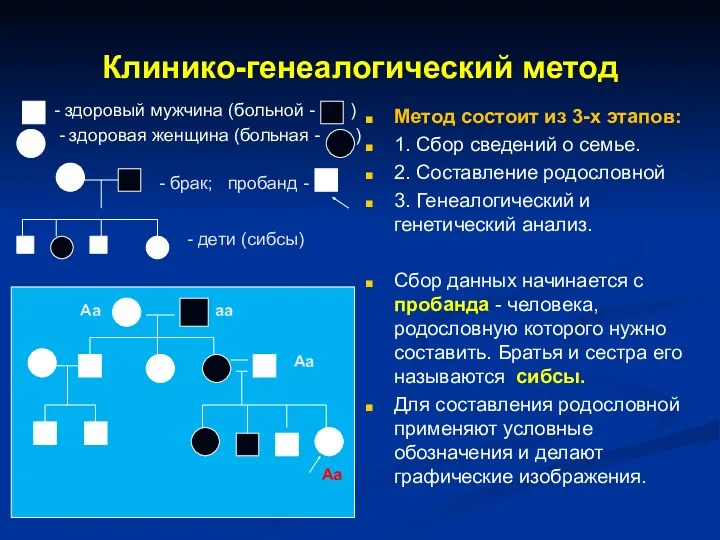
Клинико-генеалогический метод
- здоровый мужчина (больной - )
- здоровая женщина
Клинико-генеалогический метод
- здоровый мужчина (больной - )
- здоровая женщина

Близнецовый метод
Двойни встречаются 1/84 новорожденных, 1/3 из них – монозиготные (однояйцовые
Близнецовый метод
Двойни встречаются 1/84 новорожденных, 1/3 из них – монозиготные (однояйцовые

Коэффициент интеллекта, или IQ, позволяет количественно измерить генетически обусловленные умственные способности
Коэффициент интеллекта, или IQ, позволяет количественно измерить генетически обусловленные умственные способности
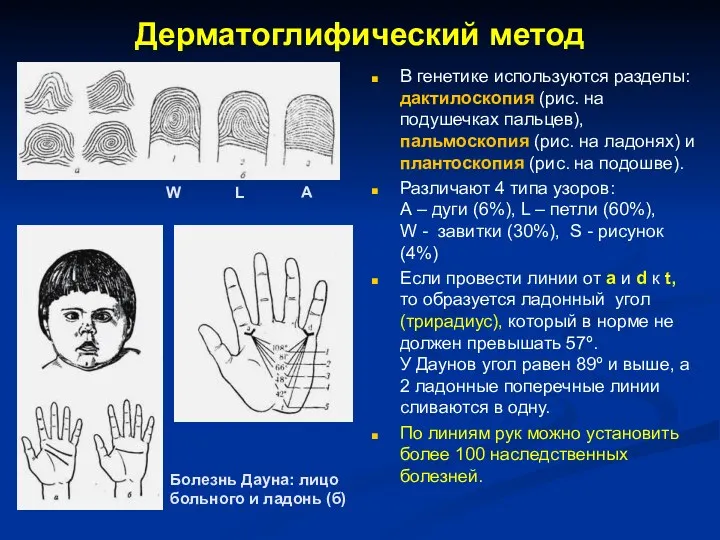
Дерматоглифический метод
В генетике используются разделы: дактилоскопия (рис. на подушечках пальцев), пальмоскопия
Дерматоглифический метод
В генетике используются разделы: дактилоскопия (рис. на подушечках пальцев), пальмоскопия

Цитогенетический метод
В 1956 г. швед. ученые Д. Тийо и А Левин
Цитогенетический метод
В 1956 г. швед. ученые Д. Тийо и А Левин
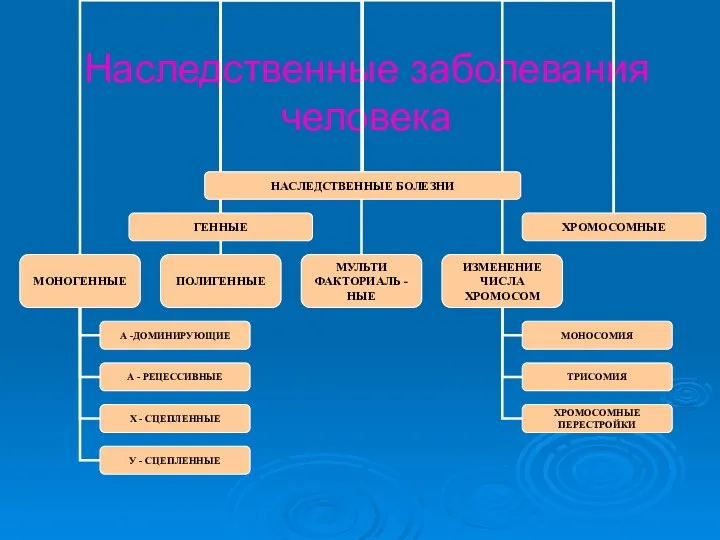
Наследственные заболевания человека
Наследственные заболевания человека

Заболевания, обусловленные изменениями числа и структуры хромосом(генные и хромосомные мутации соответственно),
Заболевания, обусловленные изменениями числа и структуры хромосом(генные и хромосомные мутации соответственно),
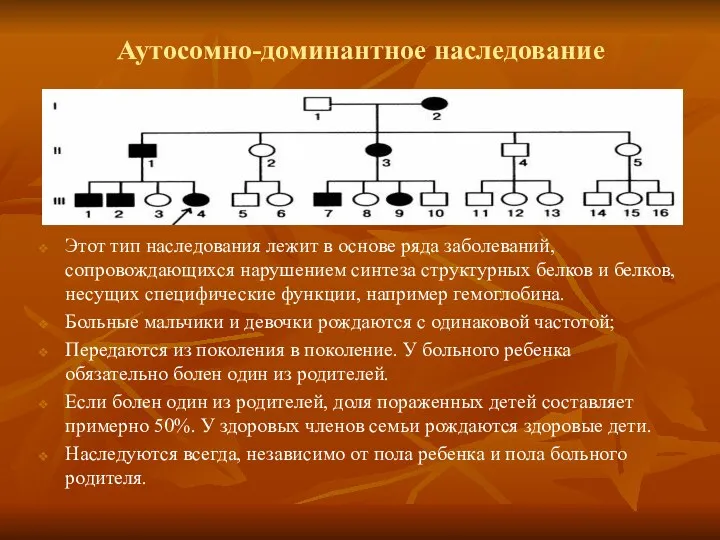
Аутосомно-доминантное наследование
Этот тип наследования лежит в основе ряда заболеваний, сопровождающихся нарушением
Аутосомно-доминантное наследование
Этот тип наследования лежит в основе ряда заболеваний, сопровождающихся нарушением

Основные понятия,используемые при характеристике заболеваний
Эпикант (эпикантус) - вертикальная складка кожи полулунной
Основные понятия,используемые при характеристике заболеваний
Эпикант (эпикантус) - вертикальная складка кожи полулунной

Акроцефалия - высокий «башенный» череп
Алопеция – стойкое или временное выпадение
Акроцефалия - высокий «башенный» череп
Алопеция – стойкое или временное выпадение

Прогерия – преждевременное старение организма
Птеригиум – крыловидные складки кожи
Птоз – опущение
Прогерия – преждевременное старение организма
Птеригиум – крыловидные складки кожи
Птоз – опущение
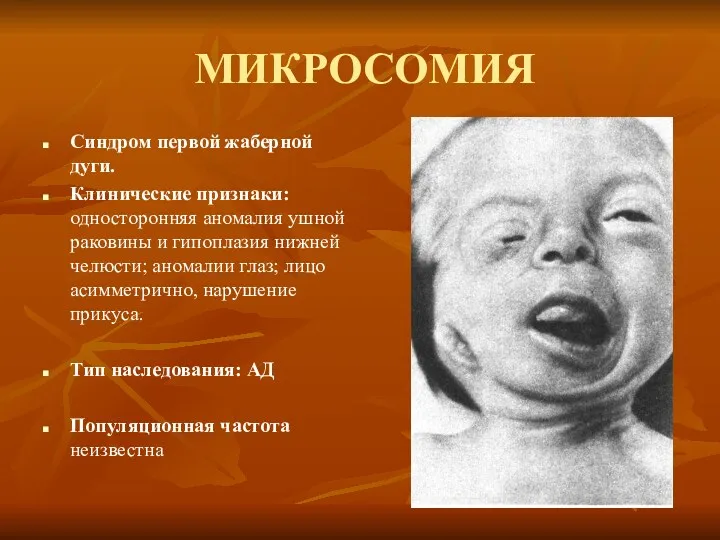
МИКРОСОМИЯ
Синдром первой жаберной дуги.
Клинические признаки: односторонняя аномалия ушной раковины и
МИКРОСОМИЯ
Синдром первой жаберной дуги.
Клинические признаки: односторонняя аномалия ушной раковины и
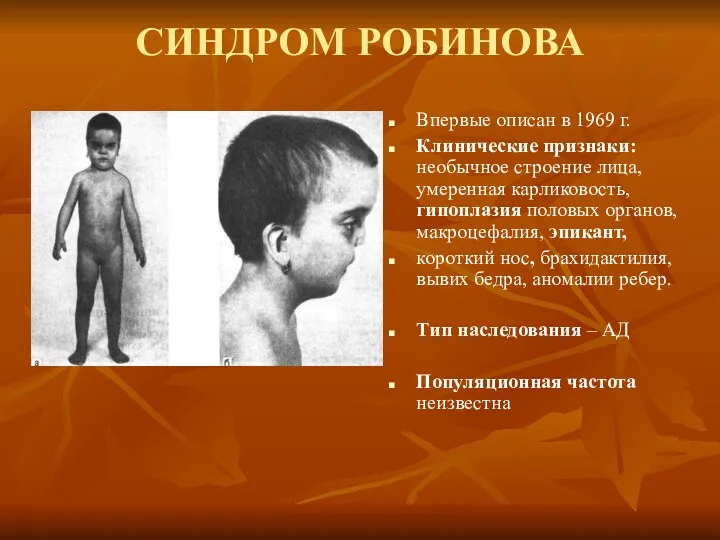
СИНДРОМ РОБИНОВА
Впервые описан в 1969 г.
Клинические признаки: необычное строение лица, умеренная
СИНДРОМ РОБИНОВА
Впервые описан в 1969 г.
Клинические признаки: необычное строение лица, умеренная
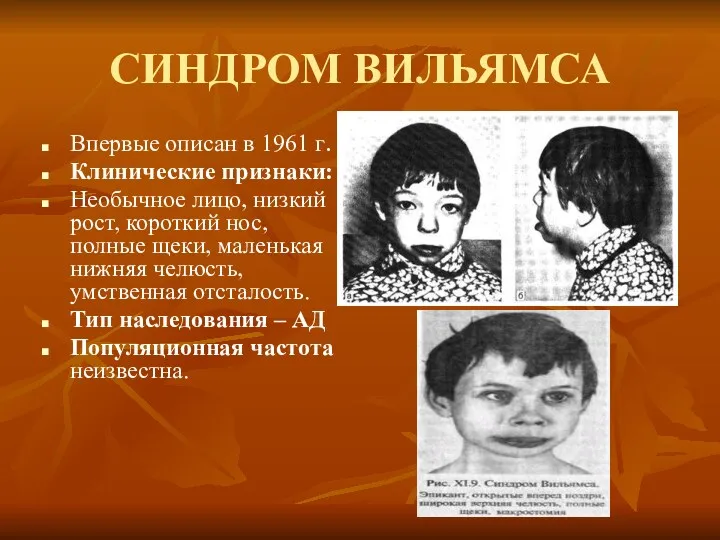
СИНДРОМ ВИЛЬЯМСА
Впервые описан в 1961 г.
Клинические признаки:
Необычное лицо, низкий рост, короткий
СИНДРОМ ВИЛЬЯМСА
Впервые описан в 1961 г.
Клинические признаки:
Необычное лицо, низкий рост, короткий
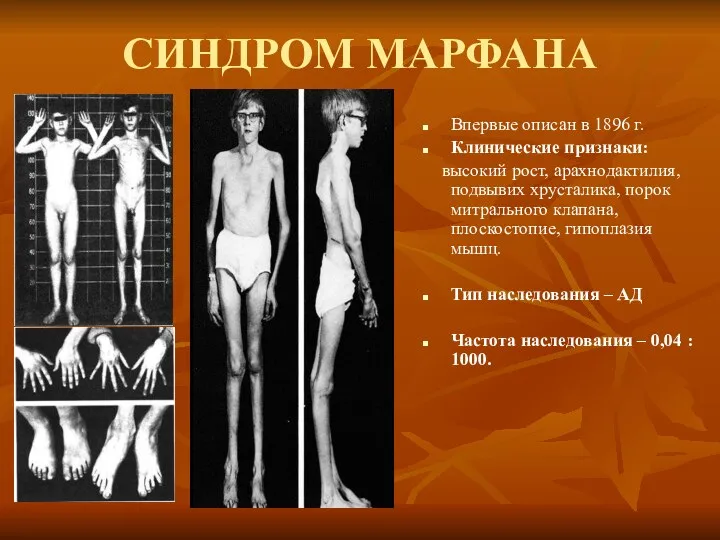
СИНДРОМ МАРФАНА
Впервые описан в 1896 г.
Клинические признаки:
высокий рост, арахнодактилия, подвывих
СИНДРОМ МАРФАНА
Впервые описан в 1896 г.
Клинические признаки:
высокий рост, арахнодактилия, подвывих

Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от
Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от
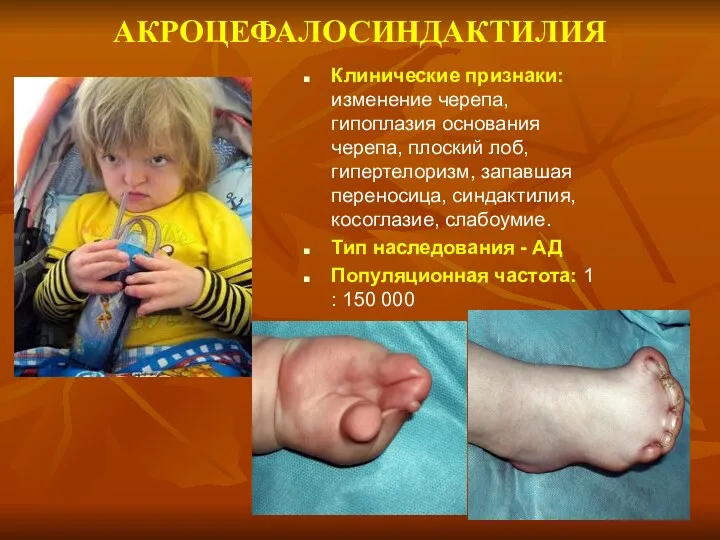
АКРОЦЕФАЛОСИНДАКТИЛИЯ
Клинические признаки: изменение черепа, гипоплазия основания черепа, плоский лоб, гипертелоризм, запавшая
АКРОЦЕФАЛОСИНДАКТИЛИЯ
Клинические признаки: изменение черепа, гипоплазия основания черепа, плоский лоб, гипертелоризм, запавшая

Трихо-рино-фалангетальный синдром
Клинические признаки: отставание в росте, лицо с грушевидным носом, оттопыренные
Трихо-рино-фалангетальный синдром
Клинические признаки: отставание в росте, лицо с грушевидным носом, оттопыренные
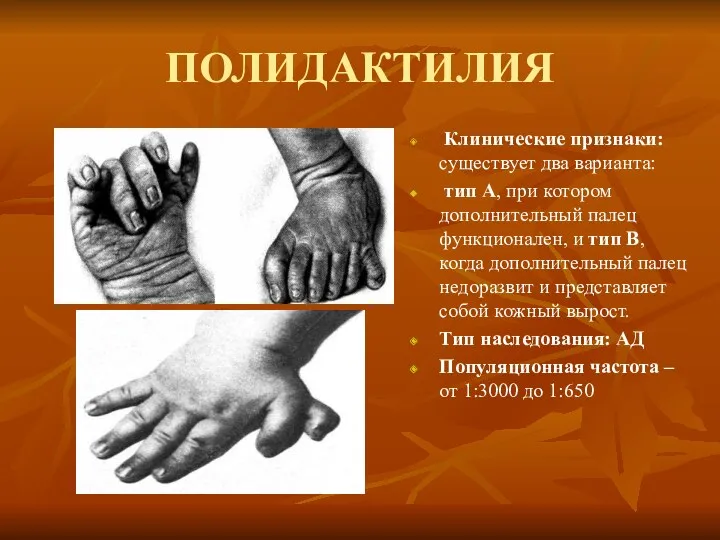
ПОЛИДАКТИЛИЯ
Клинические признаки: существует два варианта:
тип А, при котором дополнительный
ПОЛИДАКТИЛИЯ
Клинические признаки: существует два варианта:
тип А, при котором дополнительный
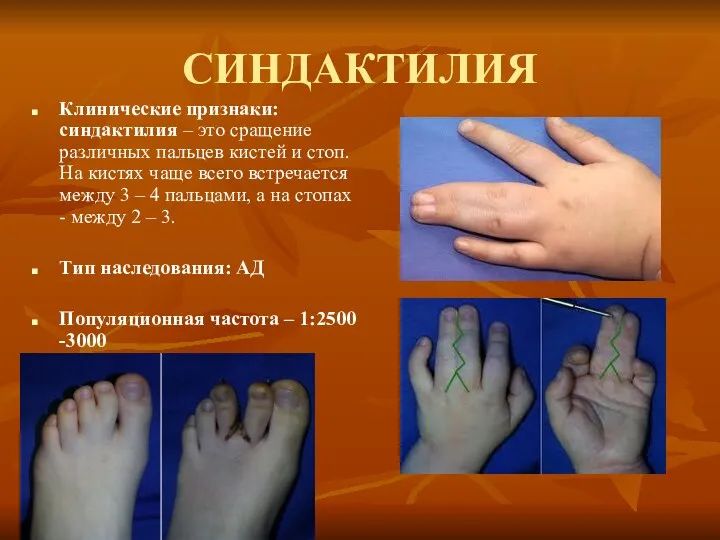
СИНДАКТИЛИЯ
Клинические признаки: синдактилия – это сращение различных пальцев кистей и стоп.
СИНДАКТИЛИЯ
Клинические признаки: синдактилия – это сращение различных пальцев кистей и стоп.
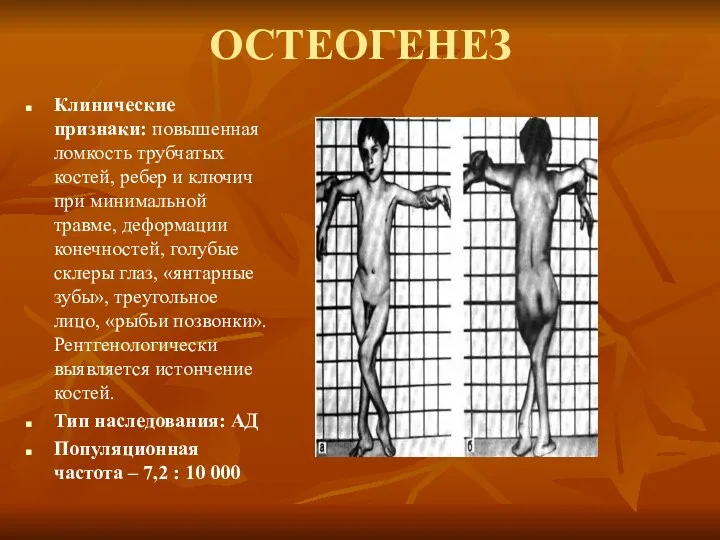
ОСТЕОГЕНЕЗ
Клинические признаки: повышенная ломкость трубчатых костей, ребер и ключич при минимальной
ОСТЕОГЕНЕЗ
Клинические признаки: повышенная ломкость трубчатых костей, ребер и ключич при минимальной
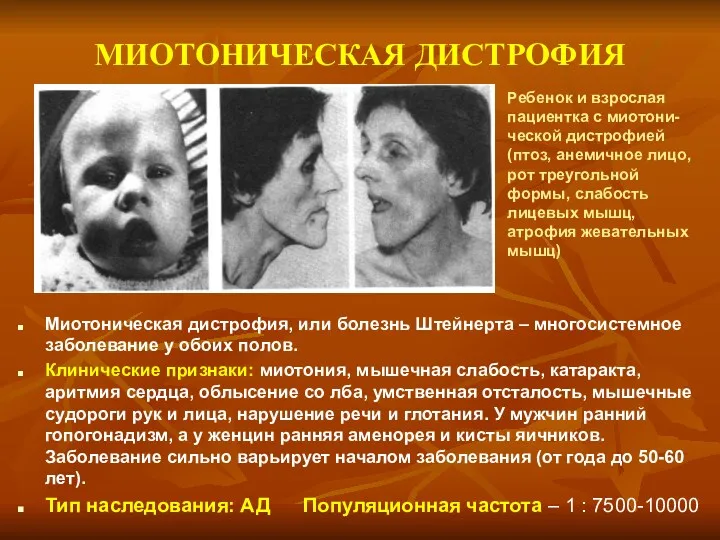
МИОТОНИЧЕСКАЯ ДИСТРОФИЯ
Миотоническая дистрофия, или болезнь Штейнерта – многосистемное заболевание у обоих
МИОТОНИЧЕСКАЯ ДИСТРОФИЯ
Миотоническая дистрофия, или болезнь Штейнерта – многосистемное заболевание у обоих
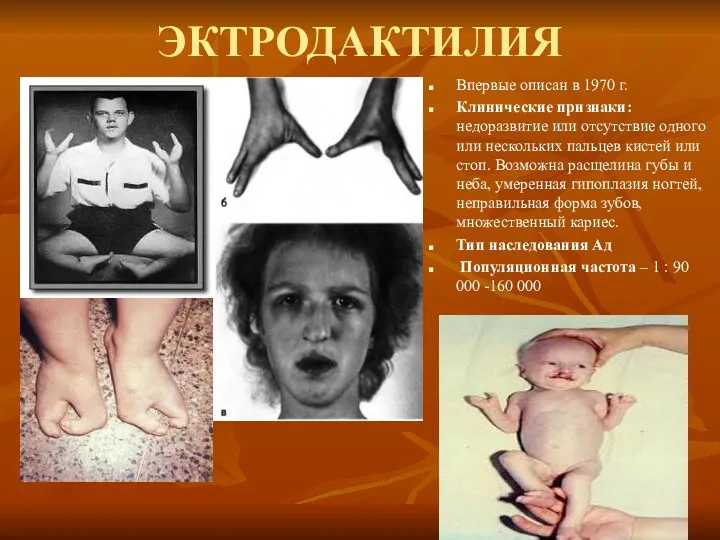
ЭКТРОДАКТИЛИЯ
Впервые описан в 1970 г.
Клинические признаки: недоразвитие или отсутствие одного или
ЭКТРОДАКТИЛИЯ
Впервые описан в 1970 г.
Клинические признаки: недоразвитие или отсутствие одного или
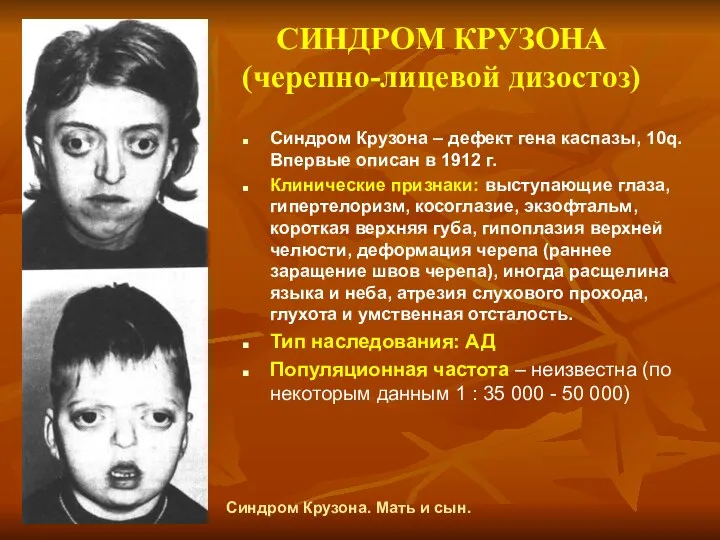
СИНДРОМ КРУЗОНА (черепно-лицевой дизостоз)
Синдром Крузона – дефект гена каспазы, 10q. Впервые
СИНДРОМ КРУЗОНА (черепно-лицевой дизостоз)
Синдром Крузона – дефект гена каспазы, 10q. Впервые

АХОНДРОПЛАЗИЯ
Клинические признаки: диспропорциональная карликовость (рост 120-130 см) за счет укорочения конечностей,
АХОНДРОПЛАЗИЯ
Клинические признаки: диспропорциональная карликовость (рост 120-130 см) за счет укорочения конечностей,
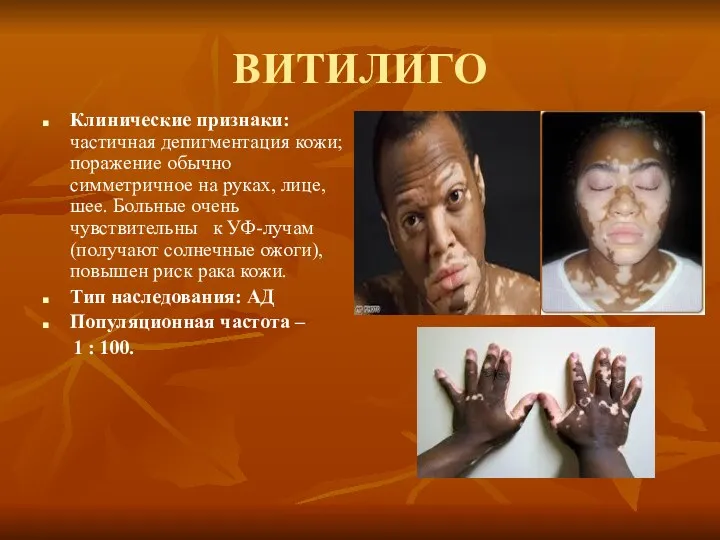
ВИТИЛИГО
Клинические признаки: частичная депигментация кожи; поражение обычно симметричное на руках, лице,
ВИТИЛИГО
Клинические признаки: частичная депигментация кожи; поражение обычно симметричное на руках, лице,

ГИПЕРТРИХОЗ («ЛЮДИ – ВОЛКИ»)
Клинические признаки: чрезмерный рост волос на всех частях
ГИПЕРТРИХОЗ («ЛЮДИ – ВОЛКИ»)
Клинические признаки: чрезмерный рост волос на всех частях

Порфирия, или вампиризм
"..Ученые выяснили, что вампиризм – это тяжелое, очень редкое
Порфирия, или вампиризм
"..Ученые выяснили, что вампиризм – это тяжелое, очень редкое
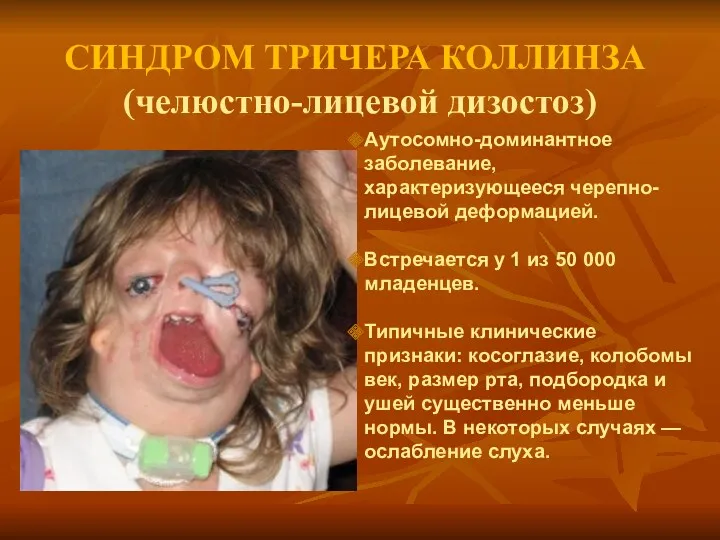
СИНДРОМ ТРИЧЕРА КОЛЛИНЗА
(челюстно-лицевой дизостоз)
Аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией.
Встречается у
СИНДРОМ ТРИЧЕРА КОЛЛИНЗА
(челюстно-лицевой дизостоз)
Аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией.
Встречается у
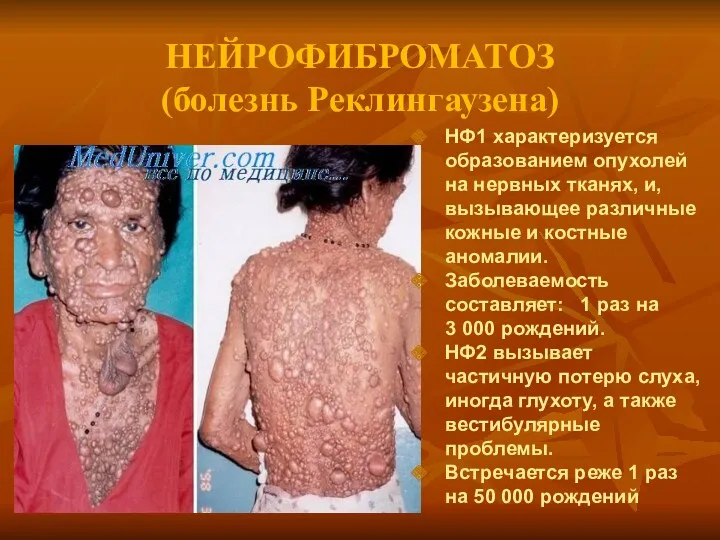
НЕЙРОФИБРОМАТОЗ
(болезнь Реклингаузена)
а
НФ1 характеризуется образованием опухолей на нервных тканях, и, вызывающее
НЕЙРОФИБРОМАТОЗ
(болезнь Реклингаузена)
а
НФ1 характеризуется образованием опухолей на нервных тканях, и, вызывающее
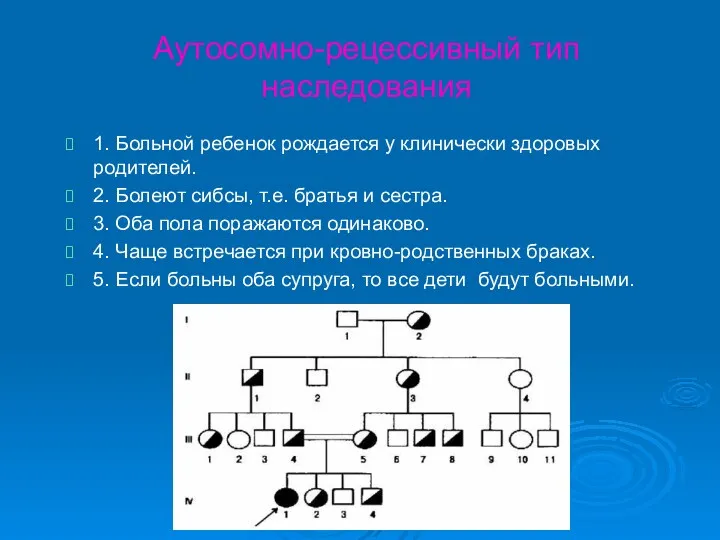
Аутосомно-рецессивный тип наследования
1. Больной ребенок рождается у клинически здоровых родителей.
2. Болеют
Аутосомно-рецессивный тип наследования
1. Больной ребенок рождается у клинически здоровых родителей.
2. Болеют
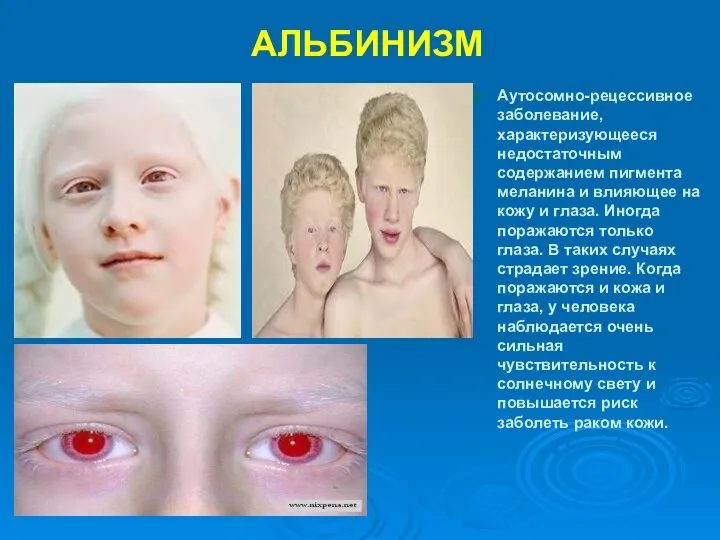
АЛЬБИНИЗМ
Аутосомно-рецессивное заболевание, характеризующееся недостаточным содержанием пигмента меланина и влияющее на кожу
АЛЬБИНИЗМ
Аутосомно-рецессивное заболевание, характеризующееся недостаточным содержанием пигмента меланина и влияющее на кожу
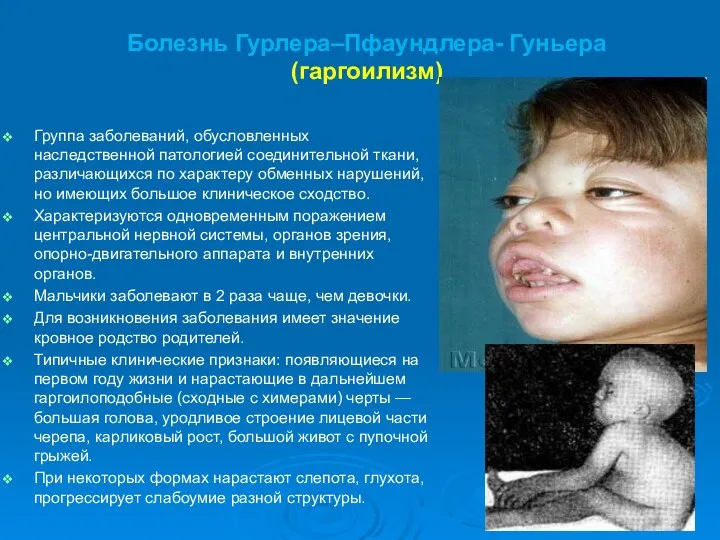
Группа заболеваний, обусловленных наследственной патологией соединительной ткани, различающихся по характеру обменных
Группа заболеваний, обусловленных наследственной патологией соединительной ткани, различающихся по характеру обменных
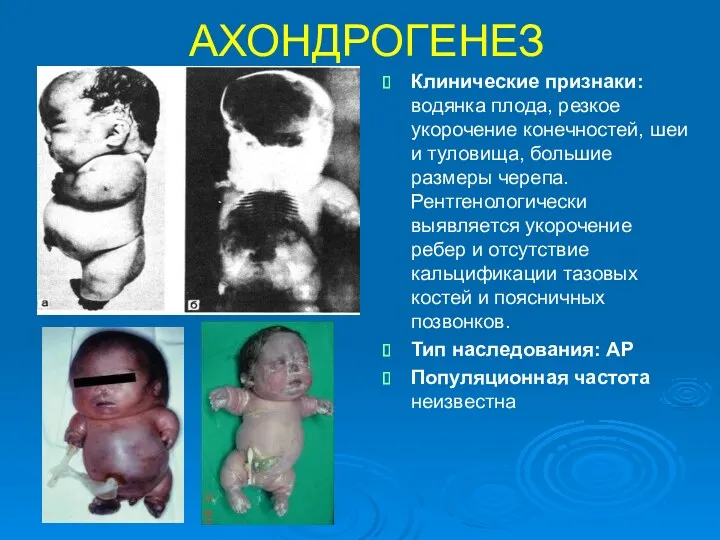
АХОНДРОГЕНЕЗ
Клинические признаки: водянка плода, резкое укорочение конечностей, шеи и туловища, большие
АХОНДРОГЕНЕЗ
Клинические признаки: водянка плода, резкое укорочение конечностей, шеи и туловища, большие

Синдром Лоуренса-Муна-Барде-Бидля
Впервые описан в 1866 г. J. Laurence и R. Moon.
Клинические
Синдром Лоуренса-Муна-Барде-Бидля
Впервые описан в 1866 г. J. Laurence и R. Moon.
Клинические
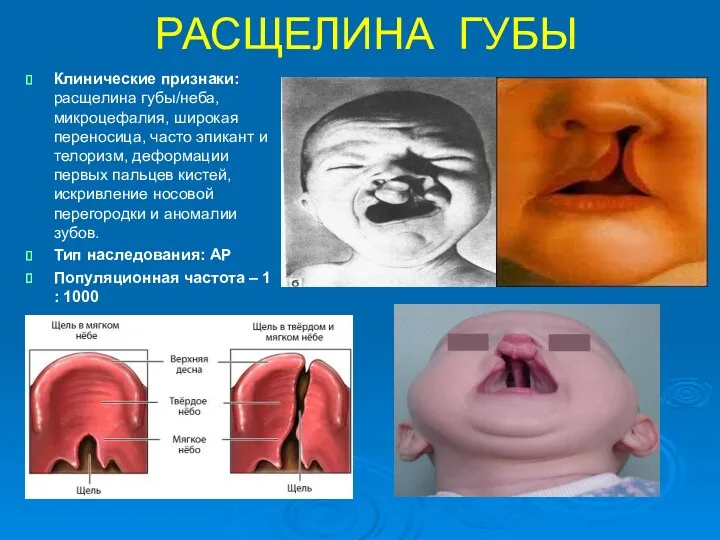
РАСЩЕЛИНА ГУБЫ
Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, часто эпикант и
РАСЩЕЛИНА ГУБЫ
Клинические признаки: расщелина губы/неба, микроцефалия, широкая переносица, часто эпикант и
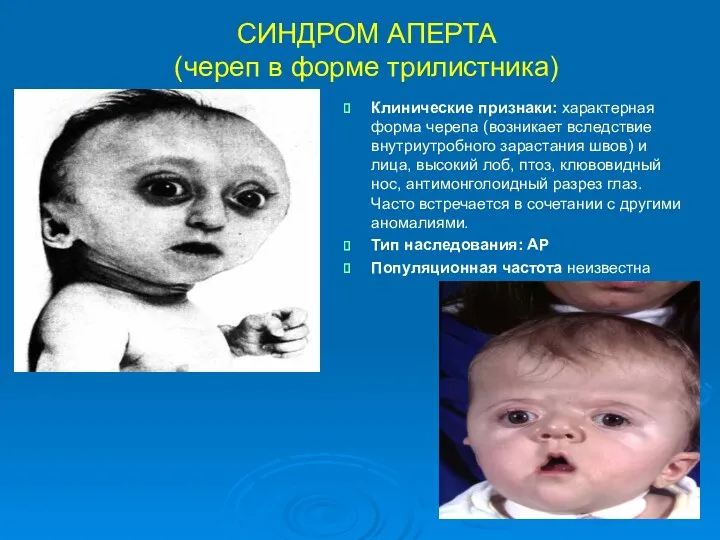
СИНДРОМ АПЕРТА
(череп в форме трилистника)
Клинические признаки: характерная форма черепа (возникает вследствие
СИНДРОМ АПЕРТА
(череп в форме трилистника)
Клинические признаки: характерная форма черепа (возникает вследствие
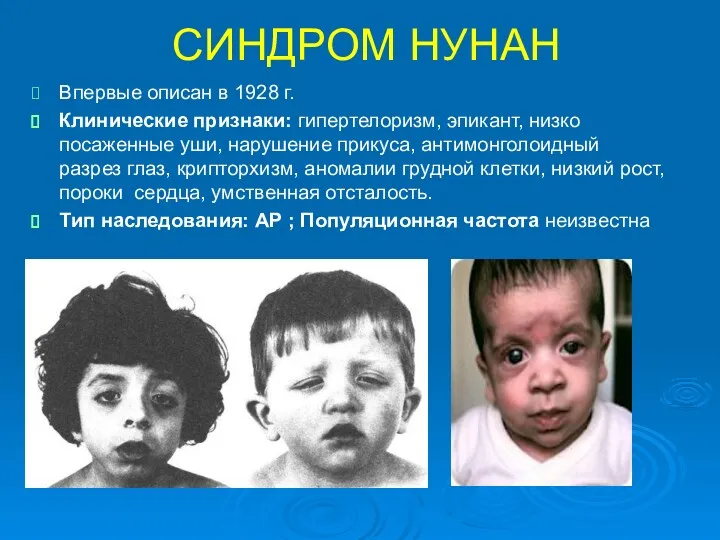
СИНДРОМ НУНАН
Впервые описан в 1928 г.
Клинические признаки: гипертелоризм, эпикант, низко
СИНДРОМ НУНАН
Впервые описан в 1928 г.
Клинические признаки: гипертелоризм, эпикант, низко

СИНДРОМ КОККЕЙНА
Впервые описан в 1946 г.
Клинические признаки: низкорослость, старообразное лицо,
СИНДРОМ КОККЕЙНА
Впервые описан в 1946 г.
Клинические признаки: низкорослость, старообразное лицо,

КСЕРОДЕРМА ПИГМЕНТНАЯ (дерматоз Капоши)
Пигментная ксеродерма – заболевание, протекающее с поражением кожи,
КСЕРОДЕРМА ПИГМЕНТНАЯ (дерматоз Капоши)
Пигментная ксеродерма – заболевание, протекающее с поражением кожи,
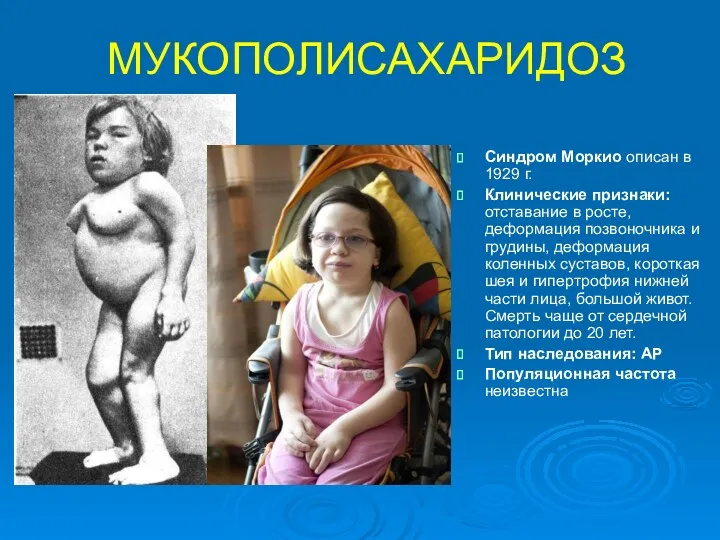
МУКОПОЛИСАХАРИДОЗ
Синдром Моркио описан в 1929 г.
Клинические признаки: отставание в росте, деформация
МУКОПОЛИСАХАРИДОЗ
Синдром Моркио описан в 1929 г.
Клинические признаки: отставание в росте, деформация

ФЕНИЛКЕТОНУРИЯ
Фенилкетонурия – болезнь аминокислотного обмена. Описана в 1934 г. А. Фелингом.
ФЕНИЛКЕТОНУРИЯ
Фенилкетонурия – болезнь аминокислотного обмена. Описана в 1934 г. А. Фелингом.

Доминантное наследование, сцепленное с X-хромосомой-этот тип наследования прослеживается, например, при фосфат-диабете.
Рецессивное
Рецессивное

Действие мутантного гена проявляется только при XY-наборе половых хромосом, т.е. у
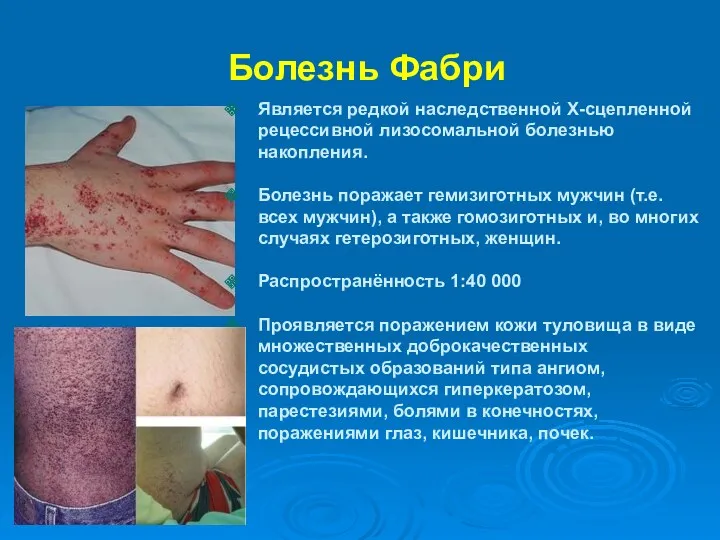
Болезнь Фабри
Является редкой наследственной Х-сцепленной рецессивной лизосомальной болезнью накопления.
Болезнь поражает гемизиготных
Болезнь Фабри
Является редкой наследственной Х-сцепленной рецессивной лизосомальной болезнью накопления.
Болезнь поражает гемизиготных

Действие доминантного мутантного гена проявляется в любом наборе половых хромосом (XX,
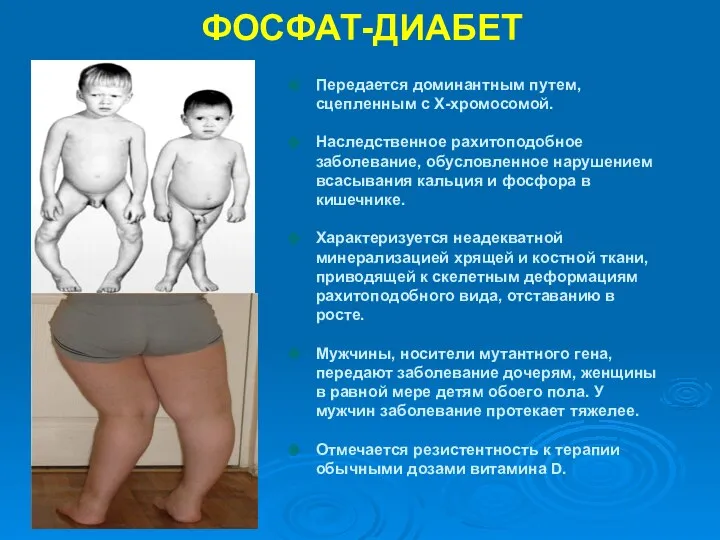
ФОСФАТ-ДИАБЕТ
Передается доминантным путем, сцепленным с Х-хромосомой.
Наследственное рахитоподобное заболевание, обусловленное нарушением
ФОСФАТ-ДИАБЕТ
Передается доминантным путем, сцепленным с Х-хромосомой.
Наследственное рахитоподобное заболевание, обусловленное нарушением

ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомные заболевания связаны с аномалиями числа или структуры хромосом.
Для них
ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомные заболевания связаны с аномалиями числа или структуры хромосом.
Для них
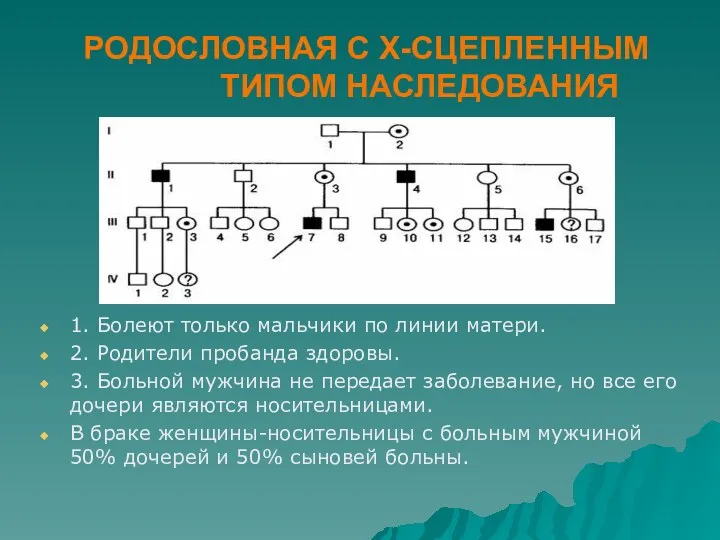
РОДОСЛОВНАЯ С Х-СЦЕПЛЕННЫМ ТИПОМ НАСЛЕДОВАНИЯ
1. Болеют только мальчики по линии
РОДОСЛОВНАЯ С Х-СЦЕПЛЕННЫМ ТИПОМ НАСЛЕДОВАНИЯ
1. Болеют только мальчики по линии
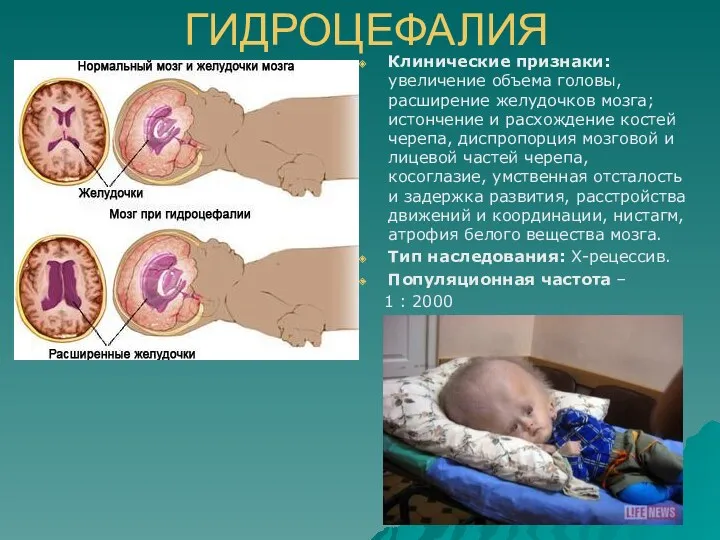
ГИДРОЦЕФАЛИЯ
Клинические признаки: увеличение объема головы, расширение желудочков мозга; истончение и расхождение
ГИДРОЦЕФАЛИЯ
Клинические признаки: увеличение объема головы, расширение желудочков мозга; истончение и расхождение
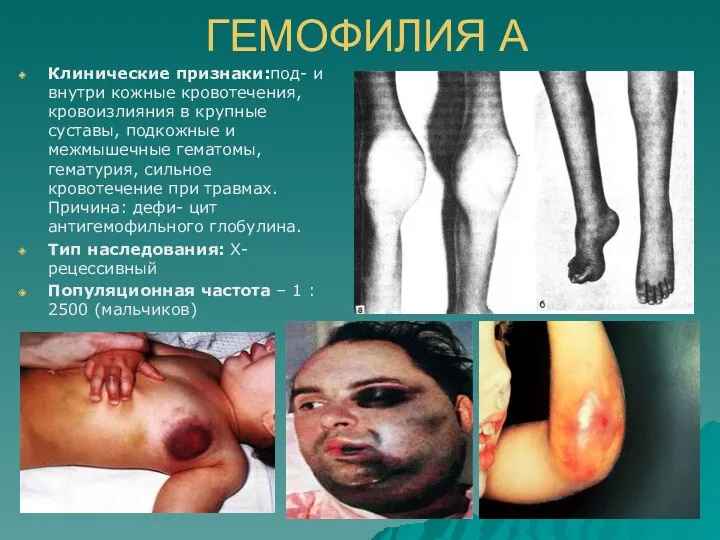
ГЕМОФИЛИЯ А
Клинические признаки:под- и внутри кожные кровотечения, кровоизлияния в крупные суставы,
ГЕМОФИЛИЯ А
Клинические признаки:под- и внутри кожные кровотечения, кровоизлияния в крупные суставы,
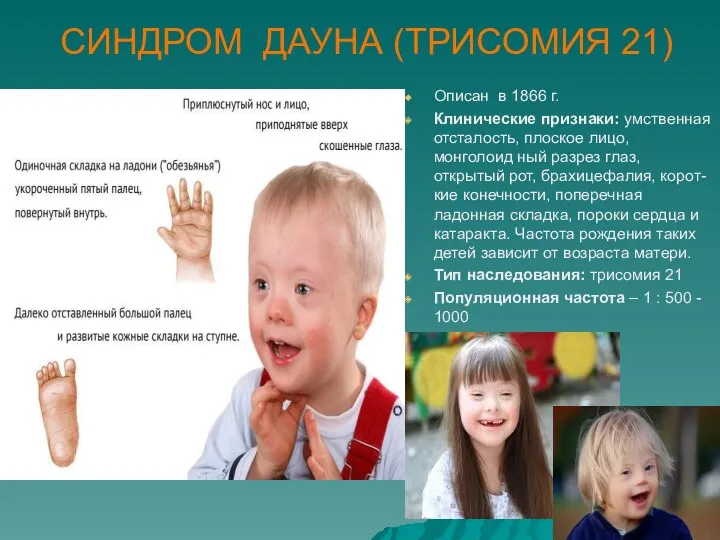
СИНДРОМ ДАУНА (ТРИСОМИЯ 21)
Описан в 1866 г.
Клинические признаки: умственная отсталость, плоское
СИНДРОМ ДАУНА (ТРИСОМИЯ 21)
Описан в 1866 г.
Клинические признаки: умственная отсталость, плоское
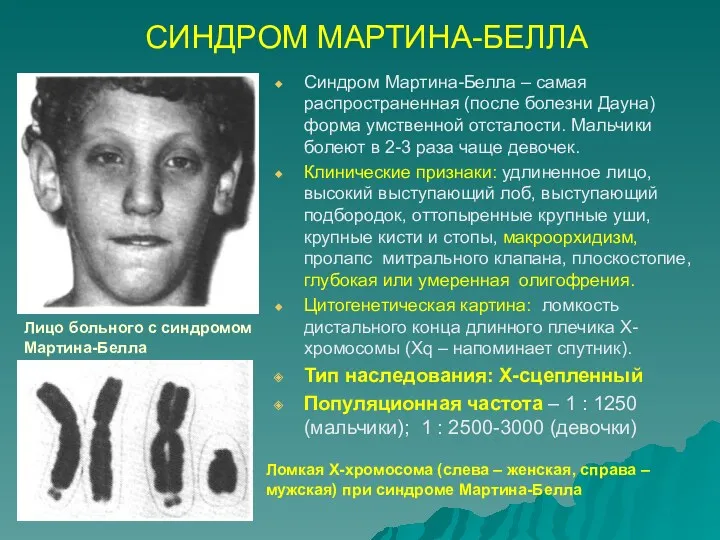
СИНДРОМ МАРТИНА-БЕЛЛА
Синдром Мартина-Белла – самая распространенная (после болезни Дауна) форма умственной
СИНДРОМ МАРТИНА-БЕЛЛА
Синдром Мартина-Белла – самая распространенная (после болезни Дауна) форма умственной

СИНДРОМ ААРСКОГО
Синдром Аарского, или лице-пальце-генитальный синдром подобно описан в 1970 г.
Клинические
СИНДРОМ ААРСКОГО
Синдром Аарского, или лице-пальце-генитальный синдром подобно описан в 1970 г.
Клинические

СИНДРОМ КОФФИНА-ЛОУРИ
Синдром впервые описан в 1966 г.
Клинические признаки: антимонголоидный разрез глаз,
СИНДРОМ КОФФИНА-ЛОУРИ
Синдром впервые описан в 1966 г.
Клинические признаки: антимонголоидный разрез глаз,
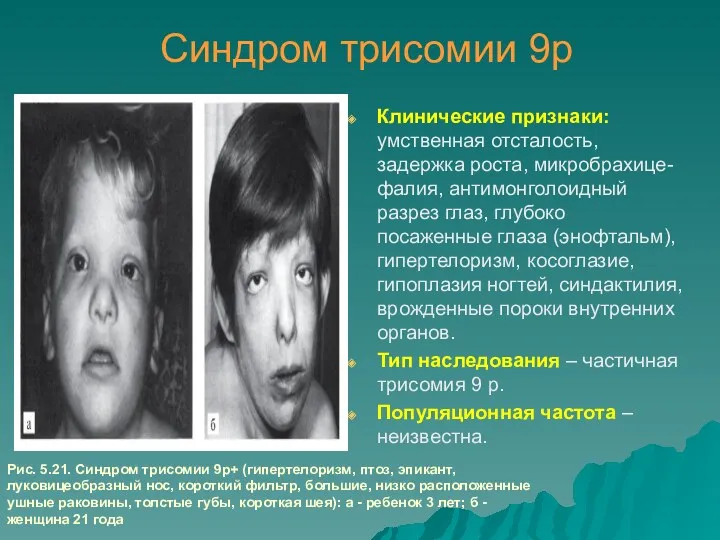
Синдром трисомии 9р
Клинические признаки: умственная отсталость, задержка роста, микробрахице-фалия, антимонголоидный разрез
Синдром трисомии 9р
Клинические признаки: умственная отсталость, задержка роста, микробрахице-фалия, антимонголоидный разрез
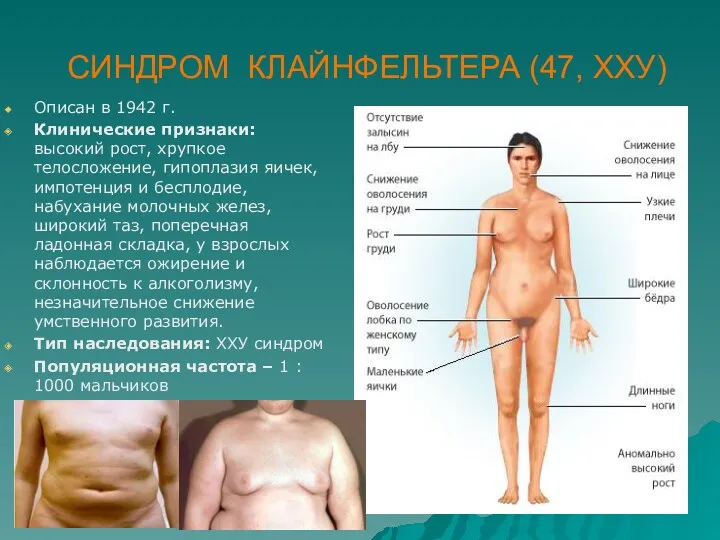
СИНДРОМ КЛАЙНФЕЛЬТЕРА (47, ХХУ)
Описан в 1942 г.
Клинические признаки: высокий рост, хрупкое
СИНДРОМ КЛАЙНФЕЛЬТЕРА (47, ХХУ)
Описан в 1942 г.
Клинические признаки: высокий рост, хрупкое
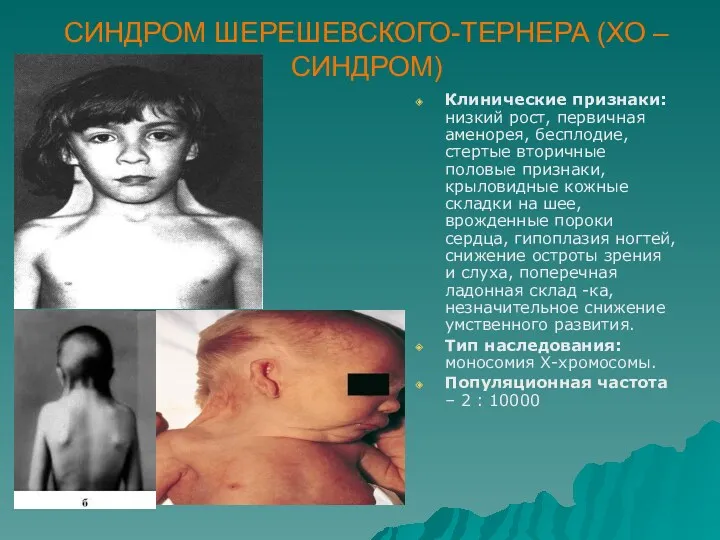
СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА (ХО –СИНДРОМ)
Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые
СИНДРОМ ШЕРЕШЕВСКОГО-ТЕРНЕРА (ХО –СИНДРОМ)
Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые

СИНДРОМ ПАТАУ (ТРИСОМИЯ 13)
Описан в 1961 г.
Клинические признаки: микроцефалия, расщепле –ние
СИНДРОМ ПАТАУ (ТРИСОМИЯ 13)
Описан в 1961 г.
Клинические признаки: микроцефалия, расщепле –ние
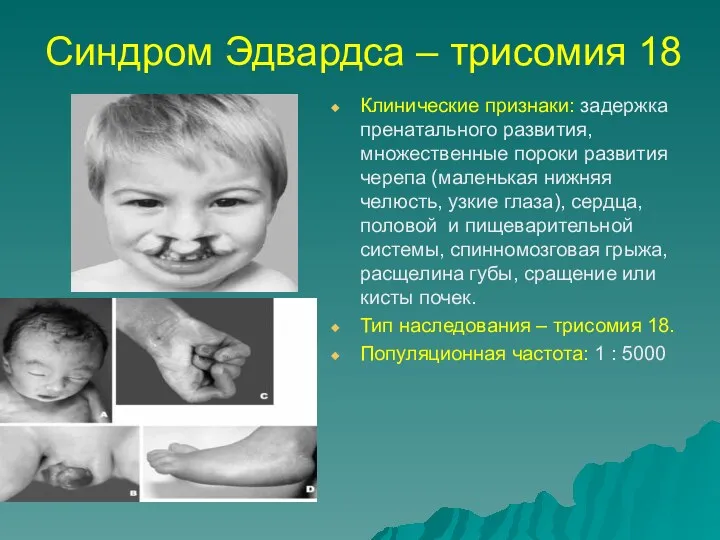
Синдром Эдвардса – трисомия 18
Клинические признаки: задержка пренатального развития, множественные пороки
Синдром Эдвардса – трисомия 18
Клинические признаки: задержка пренатального развития, множественные пороки

СИНДРОМ ЛЕЖЕНА (кошачьего крика,моносомия 5р)
Описан в 1963 г.
Клинические признаки: необычный плач,
СИНДРОМ ЛЕЖЕНА (кошачьего крика,моносомия 5р)
Описан в 1963 г.
Клинические признаки: необычный плач,

СИНДРОМ СВАЕРА (ДИСГЕНЕЗИЯ ГОНАД, ХУ ТИП )
Клинические признаки: наружные половые органы
СИНДРОМ СВАЕРА (ДИСГЕНЕЗИЯ ГОНАД, ХУ ТИП )
Клинические признаки: наружные половые органы

СИНДРОМ ЭЛЕРСА-ДАНЛОСА
Описан в 1657 г.
Клинические признаки: гиперрастяжимость соединительной ткани (нарушение синтеза
СИНДРОМ ЭЛЕРСА-ДАНЛОСА
Описан в 1657 г.
Клинические признаки: гиперрастяжимость соединительной ткани (нарушение синтеза
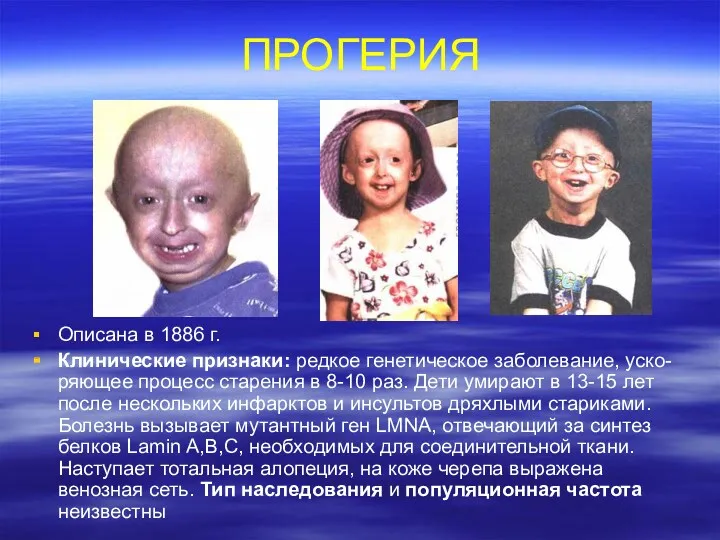
ПРОГЕРИЯ
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения
ПРОГЕРИЯ
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения

Основным способом предотвращения генетических заболеваний является тщательное клиническое обследование молодожёнов (в
Основным способом предотвращения генетических заболеваний является тщательное клиническое обследование молодожёнов (в
 Демография старения
Демография старения Инвалидность и ее причины, реабилитация
Инвалидность и ее причины, реабилитация Лечение коронавирусной инфекции Covid-19
Лечение коронавирусной инфекции Covid-19 Учение об инфекции патогенность и вирулентность микробов
Учение об инфекции патогенность и вирулентность микробов Хроническое физическое перенапряжение сердечнососудистой системы
Хроническое физическое перенапряжение сердечнососудистой системы Замещение дефектов коронки зуба коронковыми вкладками
Замещение дефектов коронки зуба коронковыми вкладками Антиангинальные средства
Антиангинальные средства Лекарственное обеспечение онкобольных
Лекарственное обеспечение онкобольных Психофизиологические основы учебного труда и интеллектуальной деятельности студентов
Психофизиологические основы учебного труда и интеллектуальной деятельности студентов Организация профилактических медицинских осмотров и диспансеризация населения
Организация профилактических медицинских осмотров и диспансеризация населения Полктың (бр) қорғаныстағы медициналық қамтамасыз етуін ұйымдастыру
Полктың (бр) қорғаныстағы медициналық қамтамасыз етуін ұйымдастыру Расстройства чувствительности, боль. Атаксии
Расстройства чувствительности, боль. Атаксии Основы биологической безопасности и защиты ПБА
Основы биологической безопасности и защиты ПБА Иммунопатология. Патологическая анатомия
Иммунопатология. Патологическая анатомия Значение белков, жиров, углеводов, витаминов и минеральных веществ в питании человека
Значение белков, жиров, углеводов, витаминов и минеральных веществ в питании человека Металлокерамика. Этапы припасовки ИК/МП на зуб
Металлокерамика. Этапы припасовки ИК/МП на зуб Беременность и ОХП. Острый панкреатит
Беременность и ОХП. Острый панкреатит Инсульт. Виды инсульта
Инсульт. Виды инсульта Группы крови животных
Группы крови животных Оперативные подходы в лечении печени
Оперативные подходы в лечении печени Беременность и новорожденный. Интереснейшие факты про беременность
Беременность и новорожденный. Интереснейшие факты про беременность Систематический обзор и мата-анализ в доказательной медицине
Систематический обзор и мата-анализ в доказательной медицине Салон красоты Все включено
Салон красоты Все включено Основы безопасности питания
Основы безопасности питания Домашний физиотерапевт FOHOW VIP
Домашний физиотерапевт FOHOW VIP Weight Loss Challenge. Группа поддержки для тех, кто хочет снизить вес
Weight Loss Challenge. Группа поддержки для тех, кто хочет снизить вес Отделение сосудистой хирургии. РСЦ Ярославской области
Отделение сосудистой хирургии. РСЦ Ярославской области Ультразвуковая диагностика заболеваний вен нижних конечностей
Ультразвуковая диагностика заболеваний вен нижних конечностей