- Генные заболевания

Содержание
- 2. Генные заболевания (ГЗ) это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на генном уровне Формы
- 3. Этиология (генные мутации, которые обусловливают наследственные заболевания) Точечные мутации – замена одного нуклеотида другим комплементарным или
- 4. Механизмы развития ГЗ (варианты первичных мутантных аллелей) Отсутствие синтеза полипептидной цепи (белка) Синтез аномальной по первичной
- 5. Варианты нарушений функций белка при ГЗ Потеря функции – за счет ингибирования процесса транскрипции/трансляции или за
- 6. Фенотипические эффекты ГЗ (клинические) Дизморфогенез (врожденные пороки развития) Нарушенный обмен веществ Смешанные эффекты
- 7. Сроки реализации патологических мутаций Внутриутробно – 25% В допубертатном периоде – 45% Пубертат, юношеский возраст –
- 8. Генокопии – заболевания со сходной клиникой, но различной генетической основой (мутации в разных локусах) Фенокопии –
- 9. Классификация генных заболеваний По генетическому принципу: Аутосомно-доминантные Аутосомно-рецессивны Х-сцепленные доминантные Х-сцепленные рецессивные У-сцепленные (голандрические) Митохондриальные
- 10. По клиническому принципу (продолжение): Нервные Нервно-мышечные Кожные Глазные Болезни опорно-двигательного аппарата и др. (в зависимости от
- 11. Патогенетический принцип (продолжение): Нарушение обмена веществ (углеводный, аминокислотный, обмен витаминов, липидов, металлов и др.) Врожденные пороки
- 12. Общие закономерности патогенеза Мутантный аллель Патологический первичный продукт Цепь последующих биохимических процессов Клетки Органы Организм
- 13. Ахондроплазия. Частота встречаемости – 1:100 000 Соотношение полов – 1:1 Критерии диагностики: Низкий рост Большой череп
- 15. Ахондроплазия (продолжение)
- 16. Синдром Марфана Частота встречаемости – 1:10 000 Соотношение полов – 1:1 75% - больны родители; 25%
- 17. Синдром Марфана Характерен клинический полиморфизм: Костно-мышечная система – арахнодактилия, высокий рост, деформации грудной клетки, гипермобильность суставов,
- 18. Синдром Марфана
- 19. Синдром Элерса-Данлоса Гетерогенное наследственное заболевание соединительной ткани с различными типами наследования АД тип – 1,7,8 АР
- 20. Критерии диагностики: Кожа – гиперрастяжимость, бархатистость, кровоточивость, стрии в области поясницы, рубцы, подкожные узелки Конечности –
- 21. Синдром Эллерса-Данлоса
- 22. Адреногенитальный синдром (врожденная гиперплазия коры надпочечников) Популяционная частота – 1:5000 новорожденных Аутосомно-рецессивное заболевание Относится к группе
- 23. Адреногенитальный синдром Сольтеряющая – полный дефицит фермента, проявляется нарушением солевого обмена (дефицит минералокортикоидов). С первых дней
- 24. Простая вирильная форма На фото представлена 42-летняя низкорослая (146 см) пациентка с адреногенитальным синдромом (АГС) с
- 25. Нарушение аминокислотного обмена - фенилкетонурия Частота встречаемости – 1:10 000, частота гетерозиготного носительства – 1:50 –
- 26. Скрининг-тесты: выявление фенилпировиноградной кислоты в моче или плазме в роддоме; с трихлорным железом)
- 27. Диагностика дети рождаются здоровыми, но с 2-х месяцев (при поступлении с молоком матери ФА) развивается клиника:
- 28. Лечение безбелковая диета с гидролизатами белка, которые не содержат ФА до 19-ти лет под контролем ФА
- 29. Целиакия. (глютеновая болезнь, болезнь Ги-Гертера-Гейбнера) – непереносимость глиадина, который содержится в клейковине Частота встречаемости – 1:3000
- 31. Врожденный гипотиреоз Частота встречаемости – 1:3500 – 1:4000 Отношение полов – 1:1 Причины: Дисгенезия щитовидной железы
- 32. Муковисцидоз (особенности наследования) Отец носитель ребенок без гена МВ носители больной Мать носитель АР тип
- 33. Патогенез обусловлен мутациями в гене CFTR, который кодирует структуру белка - трансмембранного регулятора проводимости. Ген CFTR
- 34. Форма муковисцидоза Смешанная в 75-80% Легочная 15-20% Кишечная 5% Редкие формы Ретенционная желтуха Изолированная электролитная (коллаптоидная)
- 35. Поражение органов дыхания У 1/3 больных М манифестирует на первом году жизни легочными инфекциями. Симптомы: кашель
- 36. Поражение ЖКТ Мекониальный илеус (период новорожденности) Гемолитическая анемия (в период новорожденности) Инвагинация Диарея/стеаторея Дефицит жирорастворимых витаминов
- 37. Осложнение - бесплодие Случаи полного бесплодия у женщин (например, полное отсутствие овуляции) крайне редки, чаще проблема
- 39. Генетический анализ (медико-генетическое консультирование) Пренатальная диагностика Неонатальная диагностика Постнатальная диагностика – характерные клинические симптомы, потовый тест,
- 40. Потовый тест Нужно не менее 100мг потовой жидкости, повторяется минимум трижды Начинать исследование целесообразно при достижении
- 41. Диета Основной принцип – повышение энергетической ценности пищи на 20-25%: жиры - 40-45%, белки - 15%
- 42. Заместительная терапия - ферменты Должна быть пожизненной, беспрерывной, достаточной, с физиологическим соотношением липазы, протеазы, амилазы Дозирование:
- 43. Антибактериальная терапия До 2 лет жизни ребенка наиболее частой бактериальной флорой являются St.aureus, Haemophilus influenzae. С
- 44. Ацетилцистеин (ингаляци, сироп) Амбробене (ингаляци, сироп) Рекомбинантная ДНК-аза «Пульмозим» Физические методы эвакуации мокроты Постуральный дренаж Аутогенный
- 46. Скачать презентацию

Генные заболевания (ГЗ)
это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями
Генные заболевания (ГЗ)
это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями

Этиология (генные мутации, которые обусловливают наследственные заболевания)
Точечные мутации – замена одного
Этиология (генные мутации, которые обусловливают наследственные заболевания)
Точечные мутации – замена одного

Механизмы развития ГЗ
(варианты первичных мутантных аллелей)
Отсутствие синтеза полипептидной цепи (белка)
Синтез
Механизмы развития ГЗ
(варианты первичных мутантных аллелей)
Отсутствие синтеза полипептидной цепи (белка)
Синтез

Варианты нарушений функций белка при ГЗ
Потеря функции – за счет ингибирования
Варианты нарушений функций белка при ГЗ
Потеря функции – за счет ингибирования

Фенотипические эффекты ГЗ (клинические)
Дизморфогенез (врожденные пороки развития)
Нарушенный обмен веществ
Смешанные эффекты
Фенотипические эффекты ГЗ (клинические)
Дизморфогенез (врожденные пороки развития)
Нарушенный обмен веществ
Смешанные эффекты

Сроки реализации патологических мутаций
Внутриутробно – 25%
В допубертатном периоде – 45%
Пубертат, юношеский
Сроки реализации патологических мутаций
Внутриутробно – 25%
В допубертатном периоде – 45%
Пубертат, юношеский

Генокопии – заболевания со сходной клиникой, но различной генетической основой (мутации
Генокопии – заболевания со сходной клиникой, но различной генетической основой (мутации

Классификация генных заболеваний
По генетическому принципу:
Аутосомно-доминантные
Аутосомно-рецессивны
Х-сцепленные доминантные
Х-сцепленные рецессивные
У-сцепленные (голандрические)
Митохондриальные
Классификация генных заболеваний
По генетическому принципу:
Аутосомно-доминантные
Аутосомно-рецессивны
Х-сцепленные доминантные
Х-сцепленные рецессивные
У-сцепленные (голандрические)
Митохондриальные

По клиническому принципу (продолжение):
Нервные
Нервно-мышечные
Кожные
Глазные
Болезни опорно-двигательного аппарата и др. (в зависимости от
По клиническому принципу (продолжение):
Нервные
Нервно-мышечные
Кожные
Глазные
Болезни опорно-двигательного аппарата и др. (в зависимости от

Патогенетический принцип (продолжение):
Нарушение обмена веществ (углеводный, аминокислотный, обмен витаминов, липидов, металлов
Патогенетический принцип (продолжение):
Нарушение обмена веществ (углеводный, аминокислотный, обмен витаминов, липидов, металлов

Общие закономерности патогенеза
Мутантный аллель
Патологический первичный продукт
Цепь последующих биохимических процессов
Клетки
Органы
Организм
Общие закономерности патогенеза
Мутантный аллель
Патологический первичный продукт
Цепь последующих биохимических процессов
Клетки
Органы
Организм

Ахондроплазия.
Частота встречаемости – 1:100 000
Соотношение полов – 1:1
Критерии диагностики:
Низкий рост
Большой
Ахондроплазия.
Частота встречаемости – 1:100 000
Соотношение полов – 1:1
Критерии диагностики:
Низкий рост
Большой
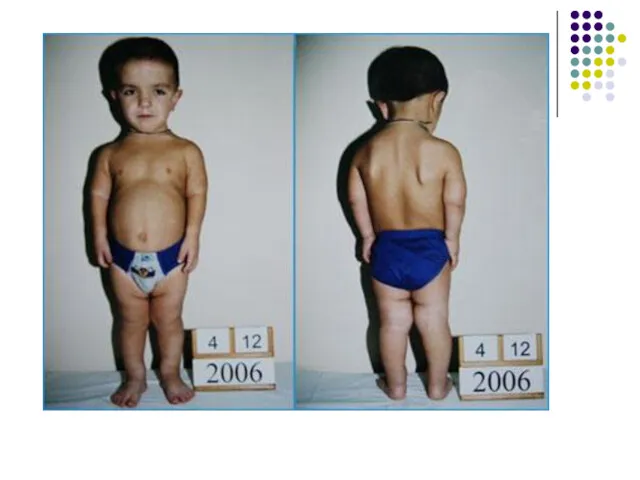

Ахондроплазия (продолжение)
Ахондроплазия (продолжение)

Синдром Марфана
Частота встречаемости – 1:10 000
Соотношение полов – 1:1
75% - больны
Синдром Марфана
Частота встречаемости – 1:10 000
Соотношение полов – 1:1
75% - больны

Синдром Марфана
Характерен клинический полиморфизм:
Костно-мышечная система – арахнодактилия, высокий рост, деформации грудной
Синдром Марфана
Характерен клинический полиморфизм:
Костно-мышечная система – арахнодактилия, высокий рост, деформации грудной
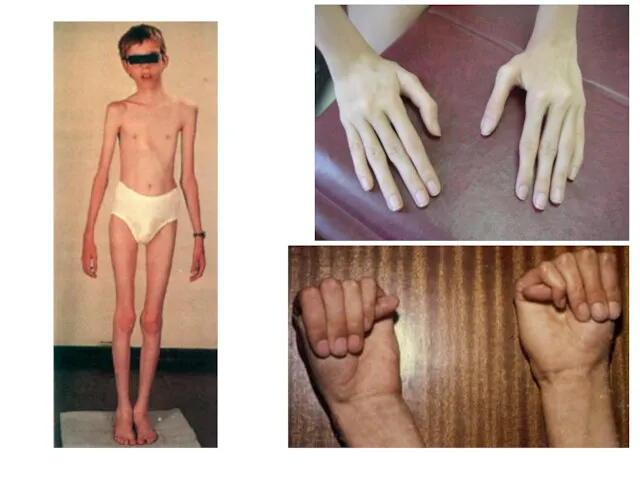
Синдром Марфана
Синдром Марфана

Синдром Элерса-Данлоса
Гетерогенное наследственное заболевание соединительной ткани с различными типами наследования
АД тип
Синдром Элерса-Данлоса
Гетерогенное наследственное заболевание соединительной ткани с различными типами наследования
АД тип

Критерии диагностики:
Кожа – гиперрастяжимость, бархатистость, кровоточивость, стрии в области поясницы, рубцы,
Критерии диагностики:
Кожа – гиперрастяжимость, бархатистость, кровоточивость, стрии в области поясницы, рубцы,
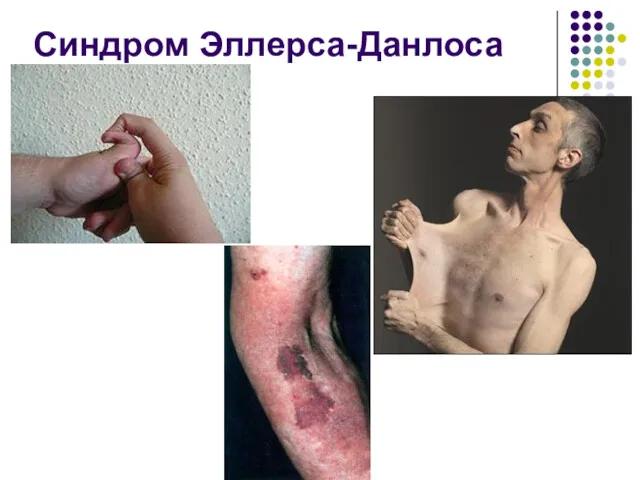
Синдром Эллерса-Данлоса
Синдром Эллерса-Данлоса

Адреногенитальный синдром (врожденная гиперплазия коры надпочечников)
Популяционная частота – 1:5000 новорожденных
Аутосомно-рецессивное заболевание
Относится
Адреногенитальный синдром (врожденная гиперплазия коры надпочечников)
Популяционная частота – 1:5000 новорожденных
Аутосомно-рецессивное заболевание
Относится

Адреногенитальный синдром
Сольтеряющая – полный дефицит фермента, проявляется нарушением солевого обмена (дефицит минералокортикоидов).
Адреногенитальный синдром
Сольтеряющая – полный дефицит фермента, проявляется нарушением солевого обмена (дефицит минералокортикоидов).
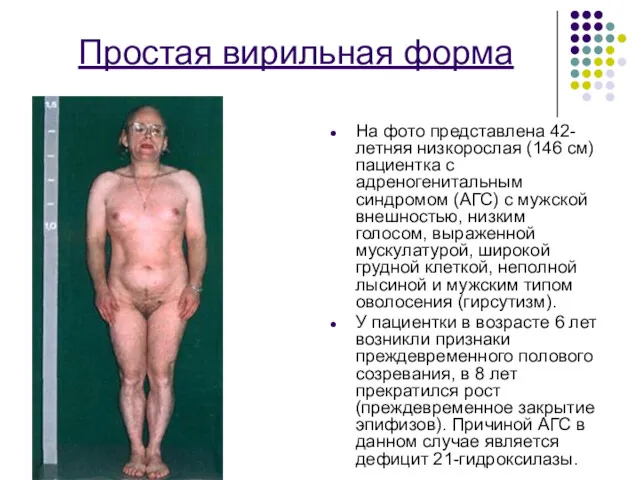
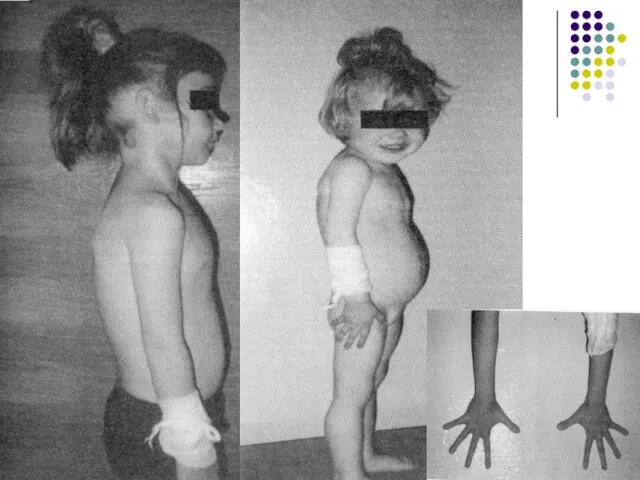
Простая вирильная форма
На фото представлена 42-летняя низкорослая (146 см) пациентка с
Простая вирильная форма
На фото представлена 42-летняя низкорослая (146 см) пациентка с

Нарушение аминокислотного обмена - фенилкетонурия
Частота встречаемости – 1:10 000, частота гетерозиготного
Нарушение аминокислотного обмена - фенилкетонурия
Частота встречаемости – 1:10 000, частота гетерозиготного
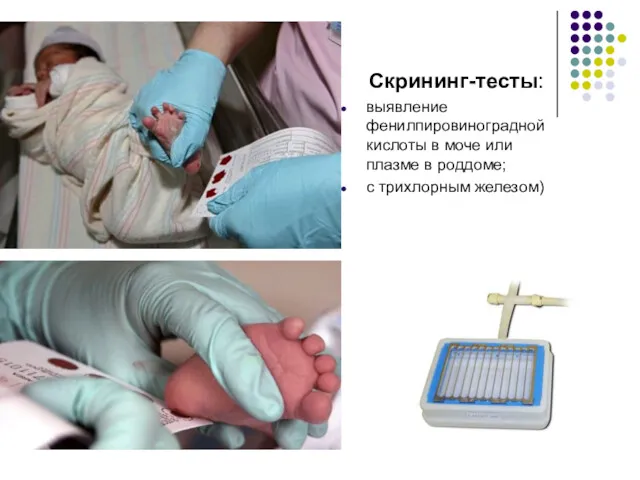
Скрининг-тесты:
выявление фенилпировиноградной кислоты в моче или плазме в роддоме;
Скрининг-тесты:
выявление фенилпировиноградной кислоты в моче или плазме в роддоме;

Диагностика
дети рождаются здоровыми, но с 2-х месяцев (при поступлении с
Диагностика
дети рождаются здоровыми, но с 2-х месяцев (при поступлении с

Лечение
безбелковая диета с гидролизатами белка, которые не содержат ФА до 19-ти
Лечение
безбелковая диета с гидролизатами белка, которые не содержат ФА до 19-ти

Целиакия.
(глютеновая болезнь, болезнь Ги-Гертера-Гейбнера) – непереносимость глиадина, который содержится в клейковине
Частота
Целиакия.
(глютеновая болезнь, болезнь Ги-Гертера-Гейбнера) – непереносимость глиадина, который содержится в клейковине
Частота
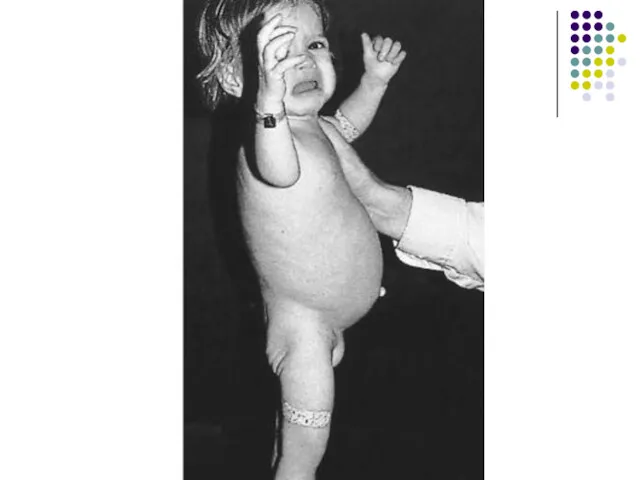

Врожденный гипотиреоз
Частота встречаемости – 1:3500 – 1:4000
Отношение полов – 1:1
Причины:
Дисгенезия
Врожденный гипотиреоз
Частота встречаемости – 1:3500 – 1:4000
Отношение полов – 1:1
Причины:
Дисгенезия
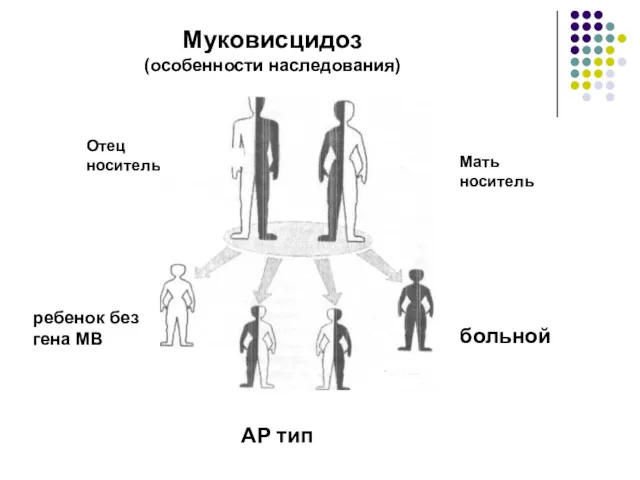
Муковисцидоз
(особенности наследования)
Отец
носитель
ребенок без
гена МВ
носители
больной
Мать
носитель
АР тип
Муковисцидоз
(особенности наследования)
Отец
носитель
ребенок без
гена МВ
носители
больной
Мать
носитель
АР тип

Патогенез обусловлен мутациями в гене CFTR, который кодирует структуру белка -
Патогенез обусловлен мутациями в гене CFTR, который кодирует структуру белка -

Форма муковисцидоза
Смешанная в 75-80%
Легочная 15-20%
Кишечная 5%
Редкие формы
Ретенционная желтуха
Изолированная электролитная (коллаптоидная)
Мекониальный илеус
Форма муковисцидоза
Смешанная в 75-80%
Легочная 15-20%
Кишечная 5%
Редкие формы
Ретенционная желтуха
Изолированная электролитная (коллаптоидная)
Мекониальный илеус

Поражение органов дыхания
У 1/3 больных М манифестирует на первом году жизни
Поражение органов дыхания
У 1/3 больных М манифестирует на первом году жизни

Поражение ЖКТ
Мекониальный илеус (период новорожденности)
Гемолитическая анемия (в период новорожденности)
Инвагинация
Диарея/стеаторея
Дефицит жирорастворимых
Поражение ЖКТ
Мекониальный илеус (период новорожденности)
Гемолитическая анемия (в период новорожденности)
Инвагинация
Диарея/стеаторея
Дефицит жирорастворимых

Осложнение - бесплодие
Случаи полного бесплодия у женщин (например, полное отсутствие овуляции)
Осложнение - бесплодие
Случаи полного бесплодия у женщин (например, полное отсутствие овуляции)

Генетический анализ (медико-генетическое консультирование)
Пренатальная диагностика
Неонатальная диагностика
Постнатальная диагностика – характерные клинические
Генетический анализ (медико-генетическое консультирование)
Пренатальная диагностика
Неонатальная диагностика
Постнатальная диагностика – характерные клинические

Потовый тест
Нужно не менее 100мг потовой жидкости, повторяется минимум трижды
Начинать исследование
Потовый тест
Нужно не менее 100мг потовой жидкости, повторяется минимум трижды
Начинать исследование

Диета
Основной принцип – повышение энергетической ценности пищи на 20-25%:
жиры -
Диета
Основной принцип – повышение энергетической ценности пищи на 20-25%:
жиры -

Заместительная терапия - ферменты
Должна быть пожизненной, беспрерывной, достаточной, с физиологическим соотношением
Заместительная терапия - ферменты
Должна быть пожизненной, беспрерывной, достаточной, с физиологическим соотношением

Антибактериальная терапия
До 2 лет жизни ребенка наиболее частой бактериальной флорой являются
Антибактериальная терапия
До 2 лет жизни ребенка наиболее частой бактериальной флорой являются

Ацетилцистеин (ингаляци, сироп)
Амбробене (ингаляци, сироп)
Рекомбинантная ДНК-аза «Пульмозим»
Физические методы эвакуации мокроты
Постуральный дренаж
Аутогенный
Ацетилцистеин (ингаляци, сироп)
Амбробене (ингаляци, сироп)
Рекомбинантная ДНК-аза «Пульмозим»
Физические методы эвакуации мокроты
Постуральный дренаж
Аутогенный
 Истинные растворы. Свойства истинных растворов. Обозначение концентраций. Способы прописывания рецептов. Общие правила
Истинные растворы. Свойства истинных растворов. Обозначение концентраций. Способы прописывания рецептов. Общие правила Лучевая диагностика заболеваний зубов, пародонта (кариес, пульпит, периодонтит, заболевания пародонта,одонтогенные кисты
Лучевая диагностика заболеваний зубов, пародонта (кариес, пульпит, периодонтит, заболевания пародонта,одонтогенные кисты Хронический холецистит
Хронический холецистит Клинический протокол и клиническое руководство по искусственному прерыванию беременности
Клинический протокол и клиническое руководство по искусственному прерыванию беременности Осуществление сестринского ухода и реабилитационные мероприятия при решении комплекса проблем пациентов с хроническим гастритом
Осуществление сестринского ухода и реабилитационные мероприятия при решении комплекса проблем пациентов с хроническим гастритом Закриті травми м’яких тканин. Переломи, вивихи
Закриті травми м’яких тканин. Переломи, вивихи Переломы костей таза
Переломы костей таза Ұйқы безі гормоны және оның зат алмасудағы мәні
Ұйқы безі гормоны және оның зат алмасудағы мәні Функциональная анатомия лимфатической системы
Функциональная анатомия лимфатической системы Флегмоны кисти
Флегмоны кисти Конституциональная физиология и психология
Конституциональная физиология и психология Язвенная болезнь желудка и двенадцатиперстной кишки
Язвенная болезнь желудка и двенадцатиперстной кишки Добровольцы-медики КФУ (Добрый мед КФУ)
Добровольцы-медики КФУ (Добрый мед КФУ) Тренинг Худеем грамотно
Тренинг Худеем грамотно Язвенная болезнь у детей
Язвенная болезнь у детей Современные репродуктивные технологии: медико-этические и социальные проблемы
Современные репродуктивные технологии: медико-этические и социальные проблемы 86861
86861 Кодирование травм и отравлений
Кодирование травм и отравлений Организация санитарно-противоэпидемического обеспечения в ЧС
Организация санитарно-противоэпидемического обеспечения в ЧС ЖИТС-пен ауыратын науқастардағы комплаенс
ЖИТС-пен ауыратын науқастардағы комплаенс Проблемы ранней беременности у подростков
Проблемы ранней беременности у подростков Мытье химической посуды. Механические и физические методы очистки, сушка химической посуды
Мытье химической посуды. Механические и физические методы очистки, сушка химической посуды Электрокардиостимуляторы – виды, показания
Электрокардиостимуляторы – виды, показания Почечная колика
Почечная колика Лабораторная диагностика инфекций, передающихся половым путем
Лабораторная диагностика инфекций, передающихся половым путем Жақ-бет аймағының терісі
Жақ-бет аймағының терісі проект
проект Виразкова хвороба шлунку та дванадцятипалої кишки
Виразкова хвороба шлунку та дванадцятипалої кишки