- Генные болезни

Содержание
- 2. Генные болезни – это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена.
- 3. Классификация аутосомно-доминантный тип аутосомно-рецессивный Х-сцепленный
- 4. Фенилкетонурия наследственная болезнь, обусловленная нарушением обмена фенилаланина; проявляется отставанием в физическом развитии и прогрессирующим слабоумием, расстройствами
- 5. Этиология и патогенез генетически детерминированный дефект фермента фенилаланин-4-гидроксилазы, который катализирует реакцию гидроксилирования L-фенилаланина в тирозин. фенилаланин
- 6. Клиника В первые недели жизни младенца заметить наличие фенилкетонурии невозможно! Через 2-6 месяцев после рождения ребенок
- 7. Диагностика Диагностика производится полуколичественным тестом или количественным определением фенилаланина в крови. При не леченных случаях возможно
- 8. Лечение Лечение фенилкетонурии проводится диетотерапией - необходимо придерживаться диеты со строгими ограничениями содержания в продуктах фенилаланина
- 9. Галактоземия наследственное заболевание, в основе которого лежит нарушение обмена веществ на пути преобразования галактозы в глюкозу.
- 10. Этиология и патогенез Галактоза, поступающая с пищей в составе молочного сахара — лактозы, подвергается превращению, но
- 11. Клиника Заболевание проявляется в первые дни и недели жизни выраженной желтухой, увеличением печени, неврологической симптоматикой (судороги,
- 12. Диагностика и дифдиагностика Позитивные пробы на сахар и обнаружение галактозы в моче в первые дни жизни,
- 13. Лечение и профилактика При подтверждении диагноза необходим перевод ребёнка на питание с исключением, главным образом, молока.
- 14. Муковисцидоз системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза и характеризующееся поражением желез внешней секреции,
- 15. Этиология и патогенез В основе заболевания лежит генная мутация. Патологический ген локализуется в середине длинного плеча
- 16. Клиническая картина Мекониевая непроходимость - данная форма заболевания обусловлена отсутствием трипсина, что приводит к скоплению в
- 17. Кишечная форма-клиническая симптоматика кишечной формы обусловлена секреторной недостаточностью желудочно-кишечного тракта. Нарушение ферментативной активности желудочно-кишечного тракта особенно
- 18. Диагностика Диагноз муковисцидоза определяется данными клинических и лабораторных методов обследования пациента. В целях ранней диагностики муковисцидоз
- 19. Лечение Диета больного муковисцидозом должна соответствовать возрасту, содержать повышенное на 10-15% количество белка и нормальное количество
- 20. Адреногенитальный синдром врождённое патологическое состояние, обусловленное дисфункцией коры надпочечников с чрезмерной секрецией андрогенов и проявляющееся признаками
- 21. Этиология и патогенез Синдром обусловлен недостаточностью одного из ферментов, необходимых для синтеза кортизола. Дефицит кортизола стимулирует
- 22. Клиническая картина Вирильная форма проявляется главным образом избытком андрогенов .У девочек часто наблюдают врождённые изменения гениталий
- 23. Диагностика В крови и моче повышены концентрации надпочечниковых андрогенов (тестостерон, андростендион, дегидроэпиандростерон) и предшественников кортизола (17-гидроксипрогестерон)
- 24. Лечение Медикаментозное лечение. Глюкокортикоиды пожизненно (подавляют гиперпродукцию АКТГ, а также надпочечнико-вых андрогенов). При натрий-дефицитной форме может
- 25. Синдром Марфана заболевание из группы наследственных коллагенопатий, заболеваний соединительной ткани человека. Заболевание наследуется по аутосомно-доминантному типу.
- 26. Этиология Синдром Марфана развивается вследствие дефекта (изменения) в гене, который определяет структуру фибрина, который играет огромную
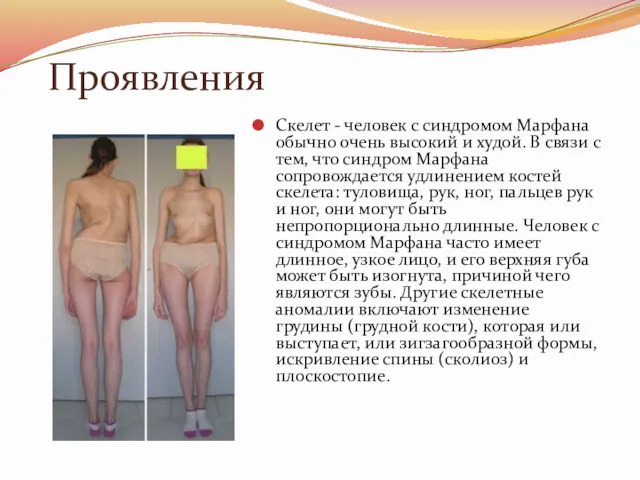
- 27. Проявления Скелет - человек с синдромом Марфана обычно очень высокий и худой. В связи с тем,
- 28. Глаза – у более, чем половины всех людей с синдромом Марфана отмечается смещение одного из двух
- 29. Сердце и кровеносные сосуды (сердечно-сосудистая система) – большинство людей с синдромом Марфана имеют аномалии, связанные с
- 30. Диагностика Нет специальных лабораторных анализов таких, как анализ крови или биопсия кожи, чтобы диагностировать синдром Марфана.
- 31. Лечение преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания
- 32. Мышечная дистрофия Дюшена наследственная прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте, симметричной атрофией мышц в
- 33. Этиология возникает в результате дефектов гена, кодирующего белок дистрофин Дистрофин локализован в плазматической мембране скелетных мышечных
- 34. Клиническая картина Мышечная дистрофия Дюшенна начинается в первые 1–3 года жизни обычно со слабости мышц тазового
- 35. Мышечный тонус обычно снижен в проксимальных группах мышц. Изменения рефлексов •• Коленные рефлексы исчезают на ранних
- 36. Диагностика и лечение Для мышечной дистрофии Дюшенна типично раннее (с 5 дня жизни) увеличение активности КФК
- 37. Мышечная дистрофия Беккера Представляет собой клинический вариант дистрофии Дюшена. Это тоже аномалия, сцепленная с Х-хромосомой и
- 38. Диагноз Основывается на характерной клинической картине, возрасте начала заболевания, семейном анамнезе и подтверждается электрофизиологическими данными, результатами
- 40. Скачать презентацию

Генные болезни – это большая группа заболеваний, возникающих в результате повреждения
Генные болезни – это большая группа заболеваний, возникающих в результате повреждения

Классификация
аутосомно-доминантный тип
аутосомно-рецессивный
Х-сцепленный
Классификация
аутосомно-доминантный тип
аутосомно-рецессивный
Х-сцепленный

Фенилкетонурия
наследственная болезнь, обусловленная нарушением обмена фенилаланина;
проявляется отставанием в физическом развитии
Фенилкетонурия
наследственная болезнь, обусловленная нарушением обмена фенилаланина;
проявляется отставанием в физическом развитии

Этиология и патогенез
генетически детерминированный дефект фермента фенилаланин-4-гидроксилазы, который катализирует реакцию
Этиология и патогенез
генетически детерминированный дефект фермента фенилаланин-4-гидроксилазы, который катализирует реакцию

Клиника
В первые недели жизни младенца заметить наличие фенилкетонурии невозможно! Через 2-6
Клиника
В первые недели жизни младенца заметить наличие фенилкетонурии невозможно! Через 2-6

Диагностика
Диагностика производится полуколичественным тестом или количественным определением фенилаланина в крови. При
Диагностика
Диагностика производится полуколичественным тестом или количественным определением фенилаланина в крови. При

Лечение
Лечение фенилкетонурии проводится диетотерапией - необходимо придерживаться диеты со строгими ограничениями
Лечение
Лечение фенилкетонурии проводится диетотерапией - необходимо придерживаться диеты со строгими ограничениями

Галактоземия
наследственное заболевание, в основе которого лежит нарушение обмена веществ на пути
Галактоземия
наследственное заболевание, в основе которого лежит нарушение обмена веществ на пути

Этиология и патогенез
Галактоза, поступающая с пищей в составе молочного сахара —
Этиология и патогенез
Галактоза, поступающая с пищей в составе молочного сахара —

Клиника
Заболевание проявляется в первые дни и недели жизни выраженной желтухой, увеличением
Клиника
Заболевание проявляется в первые дни и недели жизни выраженной желтухой, увеличением

Диагностика и дифдиагностика
Позитивные пробы на сахар и обнаружение галактозы в моче
Диагностика и дифдиагностика
Позитивные пробы на сахар и обнаружение галактозы в моче

Лечение и профилактика
При подтверждении диагноза необходим перевод ребёнка на питание с
Лечение и профилактика
При подтверждении диагноза необходим перевод ребёнка на питание с
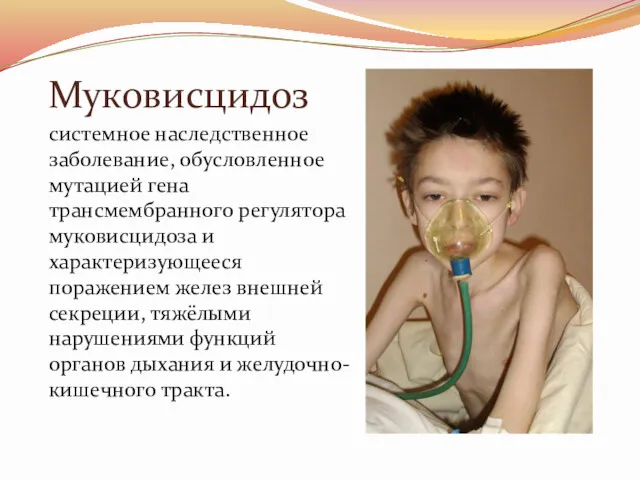
Муковисцидоз
системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза и характеризующееся
Муковисцидоз
системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза и характеризующееся

Этиология и патогенез
В основе заболевания лежит генная мутация. Патологический ген локализуется
Этиология и патогенез
В основе заболевания лежит генная мутация. Патологический ген локализуется

Клиническая картина
Мекониевая непроходимость - данная форма заболевания обусловлена отсутствием трипсина, что
Клиническая картина
Мекониевая непроходимость - данная форма заболевания обусловлена отсутствием трипсина, что

Кишечная форма-клиническая симптоматика кишечной формы обусловлена секреторной недостаточностью желудочно-кишечного тракта. Нарушение
Кишечная форма-клиническая симптоматика кишечной формы обусловлена секреторной недостаточностью желудочно-кишечного тракта. Нарушение

Диагностика
Диагноз муковисцидоза определяется данными клинических и лабораторных методов обследования пациента. В
Диагностика
Диагноз муковисцидоза определяется данными клинических и лабораторных методов обследования пациента. В

Лечение
Диета больного муковисцидозом должна соответствовать возрасту, содержать повышенное на 10-15% количество
Лечение
Диета больного муковисцидозом должна соответствовать возрасту, содержать повышенное на 10-15% количество
Адреногенитальный синдром
врождённое патологическое состояние, обусловленное дисфункцией коры надпочечников с чрезмерной секрецией
Адреногенитальный синдром
врождённое патологическое состояние, обусловленное дисфункцией коры надпочечников с чрезмерной секрецией

Этиология и патогенез
Синдром обусловлен недостаточностью одного из ферментов, необходимых для синтеза
Этиология и патогенез
Синдром обусловлен недостаточностью одного из ферментов, необходимых для синтеза

Клиническая картина
Вирильная форма проявляется главным образом избытком андрогенов .У девочек часто
Клиническая картина
Вирильная форма проявляется главным образом избытком андрогенов .У девочек часто

Диагностика
В крови и моче повышены концентрации надпочечниковых андрогенов (тестостерон, андростендион, дегидроэпиандростерон)
Диагностика
В крови и моче повышены концентрации надпочечниковых андрогенов (тестостерон, андростендион, дегидроэпиандростерон)

Лечение
Медикаментозное лечение. Глюкокортикоиды пожизненно (подавляют гиперпродукцию АКТГ, а также надпочечнико-вых андрогенов).
Лечение
Медикаментозное лечение. Глюкокортикоиды пожизненно (подавляют гиперпродукцию АКТГ, а также надпочечнико-вых андрогенов).
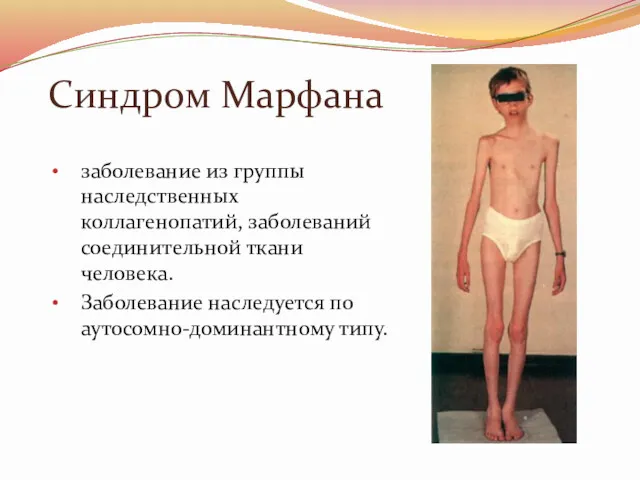
Синдром Марфана
заболевание из группы наследственных коллагенопатий, заболеваний соединительной ткани человека.
Заболевание наследуется
Синдром Марфана
заболевание из группы наследственных коллагенопатий, заболеваний соединительной ткани человека.
Заболевание наследуется
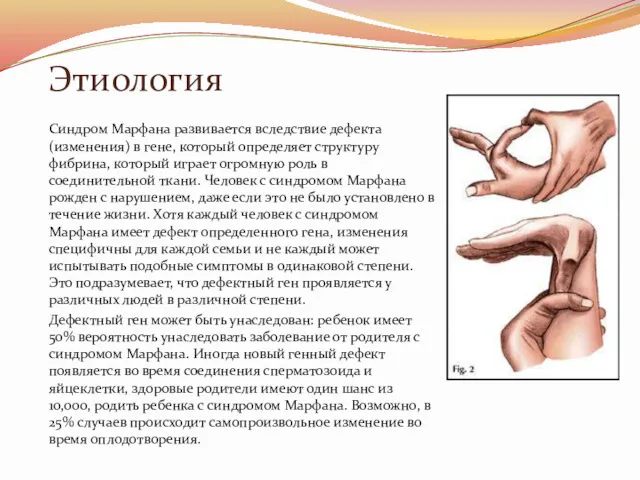
Этиология
Синдром Марфана развивается вследствие дефекта (изменения) в гене, который определяет структуру
Этиология
Синдром Марфана развивается вследствие дефекта (изменения) в гене, который определяет структуру
Проявления
Скелет - человек с синдромом Марфана обычно очень высокий и худой.
Проявления
Скелет - человек с синдромом Марфана обычно очень высокий и худой.

Глаза – у более, чем половины всех людей с синдромом Марфана
Глаза – у более, чем половины всех людей с синдромом Марфана
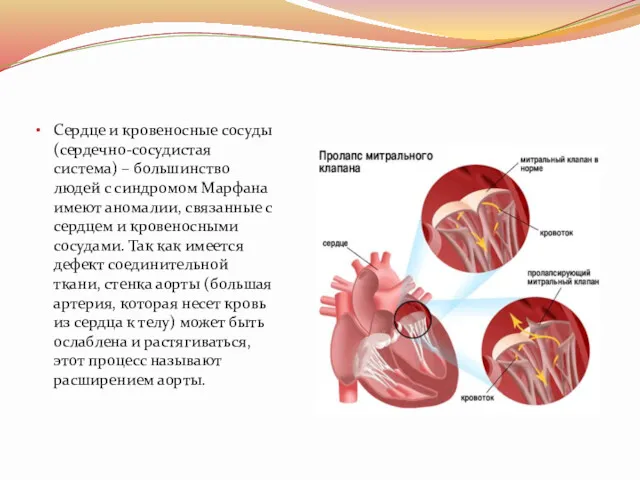
Сердце и кровеносные сосуды (сердечно-сосудистая система) – большинство людей с синдромом
Сердце и кровеносные сосуды (сердечно-сосудистая система) – большинство людей с синдромом

Диагностика
Нет специальных лабораторных анализов таких, как анализ крови или биопсия кожи,
Диагностика
Нет специальных лабораторных анализов таких, как анализ крови или биопсия кожи,

Лечение
преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания
Лечение
преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания

Мышечная дистрофия Дюшена
наследственная прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте,
Мышечная дистрофия Дюшена
наследственная прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте,

Этиология
возникает в результате дефектов гена, кодирующего белок дистрофин
Дистрофин локализован
Этиология
возникает в результате дефектов гена, кодирующего белок дистрофин
Дистрофин локализован

Клиническая картина
Мышечная дистрофия Дюшенна начинается в первые 1–3 года жизни обычно
Клиническая картина
Мышечная дистрофия Дюшенна начинается в первые 1–3 года жизни обычно

Мышечный тонус обычно снижен в проксимальных группах мышц.
Изменения рефлексов •• Коленные
Мышечный тонус обычно снижен в проксимальных группах мышц.
Изменения рефлексов •• Коленные

Диагностика и лечение
Для мышечной дистрофии Дюшенна типично раннее (с 5
Диагностика и лечение
Для мышечной дистрофии Дюшенна типично раннее (с 5

Мышечная дистрофия Беккера
Представляет собой клинический вариант дистрофии Дюшена. Это тоже аномалия,
Мышечная дистрофия Беккера
Представляет собой клинический вариант дистрофии Дюшена. Это тоже аномалия,

Диагноз
Основывается на характерной клинической картине, возрасте начала заболевания, семейном анамнезе и
Диагноз
Основывается на характерной клинической картине, возрасте начала заболевания, семейном анамнезе и
 Медицинская физиотерапевтическая аппаратура (продолжение)
Медицинская физиотерапевтическая аппаратура (продолжение) Группы крови. Переливание крови
Группы крови. Переливание крови Организмнің реактивтілігі мен резистенттілігінің патологиядағы маңызы
Организмнің реактивтілігі мен резистенттілігінің патологиядағы маңызы Междисциплинарные аспекты храпа. Влияние синдрома обструктивного апноэ сна на другие органы и системы
Междисциплинарные аспекты храпа. Влияние синдрома обструктивного апноэ сна на другие органы и системы Своя игра Мы за здоровье и безопасность
Своя игра Мы за здоровье и безопасность Тыныс алу органдарының қатерлі және қатерсіз ісіктері
Тыныс алу органдарының қатерлі және қатерсіз ісіктері Будова та розвиток центральної нервової системи
Будова та розвиток центральної нервової системи Гормоны. Функция гормонов
Гормоны. Функция гормонов Паранеопластический синдром. Общая характеристика ПНС
Паранеопластический синдром. Общая характеристика ПНС Методы визуальной диагностики заболеваний почек, мочевыводящих путей
Методы визуальной диагностики заболеваний почек, мочевыводящих путей Методика имплантации. Возможные осложнения, их профилактика и лечение
Методика имплантации. Возможные осложнения, их профилактика и лечение Методика преодоления заикания А.В. Ястребовой
Методика преодоления заикания А.В. Ястребовой ВИЧ-инфекция и туберкулез
ВИЧ-инфекция и туберкулез Патология сердечно-сосудистой системы
Патология сердечно-сосудистой системы Зертханалық жануарларды экспериментальды жолмен жұқтыру
Зертханалық жануарларды экспериментальды жолмен жұқтыру Клиническая энзимология
Клиническая энзимология Медико-биологиялық ақпаратты алу, тіркеу және жеткізудің құрылымдық сызбасы
Медико-биологиялық ақпаратты алу, тіркеу және жеткізудің құрылымдық сызбасы Сенсоневральная тугоухость. Этиология. Патогенез. Методы обследования
Сенсоневральная тугоухость. Этиология. Патогенез. Методы обследования Синдром диабетической стопы
Синдром диабетической стопы Аналіз впливу спортивного харчування на здоров'я людини
Аналіз впливу спортивного харчування на здоров'я людини Митохондриялыќ аурулар
Митохондриялыќ аурулар Желшешек
Желшешек Инфекция. Основные этапы инфекционного процесса
Инфекция. Основные этапы инфекционного процесса Тістем және оның түрлері
Тістем және оның түрлері Причины и предрасполагающие факторы к возникновению рахита
Причины и предрасполагающие факторы к возникновению рахита Ревматическая лихорадка
Ревматическая лихорадка Общие вопросы хирургической инфекции
Общие вопросы хирургической инфекции Классификация, патогенез, клиника, интенсивная терапия
Классификация, патогенез, клиника, интенсивная терапия