- Хронические лимфопролиферативные заболевания. Опухоли из зрелых В-клеток. Лимфомы

Содержание
- 2. Хронические лимфопролиферативные заболевания (ХЛПЗ) Хронические лимфопролиферативные заболевания (ХЛПЗ) - группа заболеваний, вызванная злокачественной трансформацией зрелых лимфоцитов,
- 3. Хронический лимфолейкоз (ХЛЛ) Злокачественное клональное лимфопролиферативное заболевание, характеризующееся накоплением атипичных зрелых В-лимфоцитов преимущественно в крови, костном
- 4. Хронический лимфолейкоз (ХЛЛ) Эпидемиология Хронический лимфолейкоз - самый распространенный вид хронического лейкоза и составляет около 30%
- 5. Клинические проявления Заболевание выявляется случайно. При прогрессировании опухоли наиболее частыми клиническими симптомами являются лимфаденопатия, гепатомегалия, спленомегалия,
- 6. Хронический лимфолейкоз (ХЛЛ) Картина периферической крови представлена нормальным или незначительно повышенным количеством лейкоцитов. Анемия и тромбоцитопения,
- 7. Хронический лимфолейкоз
- 10. Волосатоклеточный лейкоз Волосато-клеточный лейкоз – редкий вариант хронического лейкоза из хорошо дифференцированных В-лимфоцитов. Заболевание составляет 2%
- 11. Диагностика Костный мозг нормо- или гиперклеточный с диффузной лимфоидной инфильтрацией, часто развивается фиброз, процент «волосатых» клеток
- 12. Почему данную форму лейкоза назвали волосатоклеточной? Такое название происходит от «рваных» или «волосатых» краев раковой клетки,
- 13. Парапротеинемические гемобластозы. - опухоли системы В - лимфоцитов, дифференцирующиеся до стадии секреции иммуноглобулинов и продуцирующих в
- 14. Схема кроветворения
- 15. Парапротеинемические гемобластозы. Патогенез. Основной отличительной особенностью этих заболеваний является продукция моноклонального иммуноглобулина (М-компонент, М-градиент, М-протеин, парапротеин),
- 16. Y Y Y Y Антиген Мутация Y Y Y Y Y Y Y Y Y Y
- 17. Парапротеинемические гемобластозы Основную часть составляет IgG. При миеломе у большинства больных моноклональный синтез тяжелых цепей сочетается
- 18. Лабораторная диагностика Парапротеинемических гемобластозов. 1) повышение СОЭ, которое является результатом увеличения глобулиновой фракции крови за счет
- 19. Лабораторная диагностика парапротеинов. Разделение растворенных белков под действием постоянного электрического поля - белковый электрофорез. Электрофоретическое разделение
- 21. Лабораторная диагностика парапротеинов. Норма Патология
- 23. Парапротеинемические гемобластозы. Классификация. Миеломная болезнь (плазмоцитома, множественная миелома ) — В-клеточное лимфопролиферативное заболевание, характеризующееся клональной пролиферацией
- 24. Миеломная болезнь (множественная миелома) Миеломная болезнь — В-клеточное лимфопролиферативное заболевание, характеризующееся клональной пролиферацией в костном мозге,
- 25. Этиология 1. Ионизирующая радиация 2. Генетическая предрасположенность к развитию МБ 3. Цитогенетические нарушения. Мутация супрессорных генов
- 26. Иммунохимические варианты множественной миеломы 1. G-миелома 55-65% 2. А-миелома 20-25% 3. D-миелома 2-5% 4. Е-миелома 5.
- 27. Миеломная болезнь (множественная миелома) Клиническая картина: остеодеструкция плоских костей, полинейропатии, миеломная нефропатия с развитием почечной недостаточности,
- 28. Диагностика В костном мозге при миеломной болезни отмечается плазмоклеточная инфильтрация с анизоцитозом клеток и их ядер,
- 29. Миеломная болезнь (множественная миелома)
- 30. Миеломная болезнь (множественная миелома) До настоящего времени множественная миелома остается заболеванием, трудным для лечения, так как
- 31. Макроглобулинемия Вальденстрема Макроглобулинемия Вальденстрема — В-клеточная опухоль, морфологически представленная лимфоцитами, зрелыми плазматическими клетками и переходными формами
- 32. Макроглобулинемия Вальденстрема Формы течения: бессимптомная, медленно прогрессирующая (продолжительность жизни более 5 лет), быстро прогрессирующая (длительность жизни
- 33. Клинические проявления МВ. Обусловлены пролиферацией лимфоидных элементов в костном мозге, печени, селезенке, лимфоузлах и накоплением в
- 34. Макроглобулинемия Вальденстрема Диагностика В костном мозге отмечаются пролиферация лимфоцитов, иногда с плазматизированной цитоплазмой, увеличение плазматических клеток
- 35. Морфологическое доказательство преимущественно костномозгового лимфопролиферативного процесса Выявление моноклоновой макроглобулинемии типа IgM (не менее 10-15% от общего
- 36. Лимфомы Лимфома — группа гематологических заболеваний лимфатической ткани, характеризующихся увеличением лимфатических узлов и/или поражением различных внутренних
- 37. Лимфомы Термином неходжкинские лимфомы обозначают довольно большую группу лимфом, которые не являются болезнью Ходжкина (лимфогранулематозом). Решение
- 38. Болезнь Ходжкина (лимфогранулематоз, ЛГМ) Злокачественное заболевание лимфоидной ткани, характерным признаком которого является наличие гигантских клеток Рид-Березовского-Штернберга
- 39. Болезнь Ходжкина (лимфогранулематоз, ЛГМ) Патогенез. Опухолевым субстратом ЛГМ являются специфические гигантские клетки с дольчатым ядром и
- 40. Неходжкинские лимфомы Неходжкинские лимфомы являются гетерогенной группой неопластических заболеваний, происходящих из иммунной системы. Характеристикой лимфомы являются
- 41. Этиология неходжкинских лимфом Инфекционные Вирус Эпштейна-Барр — ассоциирован с лимфомой Бёркитта, лимфогранулематозом, фолликулярной дендритно-клеточной саркомой, экстранодальной
- 42. Неходжкинские лимфомы Клиническая картина. Наиболее часто в дебюте заболевания появляется опухоль лимфатического узла или любой другой
- 43. Неходжкинские лимфомы Общие симптомы: высокая температура (выше 38°C), причина её появления непонятна [симптом "В"] ночное потение
- 44. Неходжкинские лимфомы Диагностика. Диагноз лимфомы основывается на исследовании морфологического субстрата опухоли. Обычно исходной точкой диагностического поиска
- 45. Неходжкинские лимфомы Основной метод диагностики лимфомы – исследование поражённого лимфоузла (лимфатические узлы) или образца любой другой
- 46. Стадирование лимфом
- 47. Неходжкинские лимфомы
- 48. Фолликулярная лимфома Иммунофенотип клеток CD19+, CD10+, CD20+, CD5-, Генетика t(14;18)(q32;q21) химерный онкоген IGH/BCL2 80-90% фолликулярной лимфомы,
- 49. Фолликулярная лимфома t(14;18)(q32;q21) химерный онкоген IGH/BCL2 80-90% фолликулярной лимфомы, 30% диффузной В-крупноклеточной лимфомы Leukemia (2003) 17,
- 50. Диффузная B-крупноклеточная лимфома (ДКБЛ) Агрессивная лимфома, 40% всех неходжкинских лимфом взрослых Лечатся плохо Иммунофенотип CD19+; CD22+;
- 51. Лимфома Беркитта ЛБ ‑ опухоль из клеток герминативного центра фолликулов со зрелым В-кле-точным иммунофенотипом. Самая быстрорастущая
- 52. t(8;14)(q24;q32) химерный онкоген IGH/MYC Встречается в 70% лимфомы Беркитта c-Myc IgH 1 – маркер молекулярного веса
- 53. Грибовидный микоз/синдром Сезари (первичная Т-клеточная кожная лимфома) Составляет 2-3% всех злокачественных лимфом. Заболевание развивается медленно. Характерно
- 55. Скачать презентацию

Хронические лимфопролиферативные заболевания (ХЛПЗ)
Хронические лимфопролиферативные заболевания (ХЛПЗ) - группа заболеваний, вызванная
Хронические лимфопролиферативные заболевания (ХЛПЗ)
Хронические лимфопролиферативные заболевания (ХЛПЗ) - группа заболеваний, вызванная
Хронический лимфолейкоз (ХЛЛ)
Злокачественное клональное лимфопролиферативное заболевание, характеризующееся накоплением атипичных зрелых В-лимфоцитов
Хронический лимфолейкоз (ХЛЛ)
Злокачественное клональное лимфопролиферативное заболевание, характеризующееся накоплением атипичных зрелых В-лимфоцитов

Хронический лимфолейкоз (ХЛЛ)
Эпидемиология
Хронический лимфолейкоз - самый распространенный вид хронического лейкоза и
Хронический лимфолейкоз (ХЛЛ)
Эпидемиология
Хронический лимфолейкоз - самый распространенный вид хронического лейкоза и

Клинические проявления
Заболевание выявляется случайно. При прогрессировании опухоли наиболее частыми клиническими симптомами
Клинические проявления Заболевание выявляется случайно. При прогрессировании опухоли наиболее частыми клиническими симптомами

Хронический лимфолейкоз (ХЛЛ)
Картина периферической крови
представлена нормальным или незначительно повышенным количеством
Хронический лимфолейкоз (ХЛЛ)
Картина периферической крови
представлена нормальным или незначительно повышенным количеством
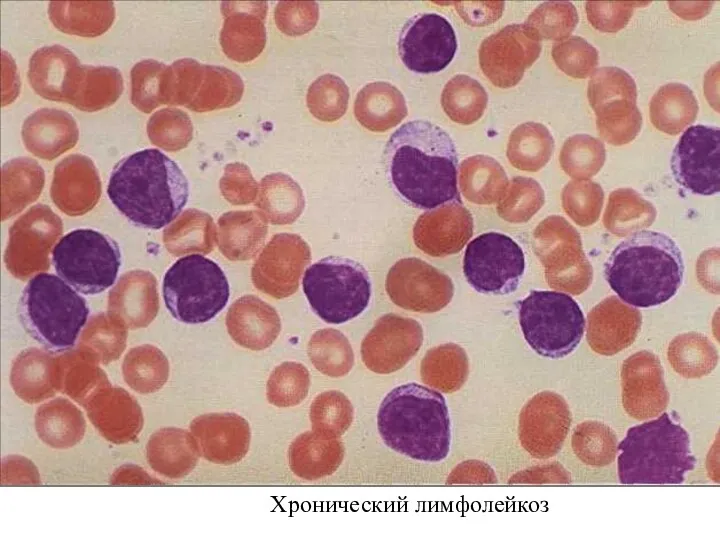
Хронический лимфолейкоз
Хронический лимфолейкоз
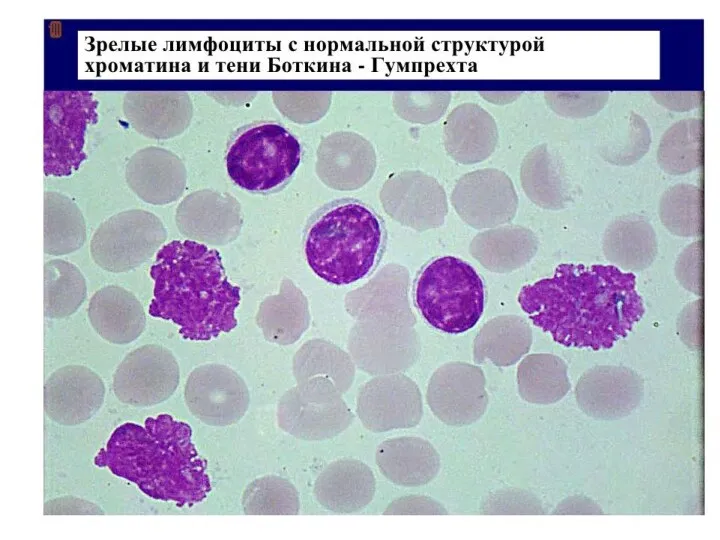
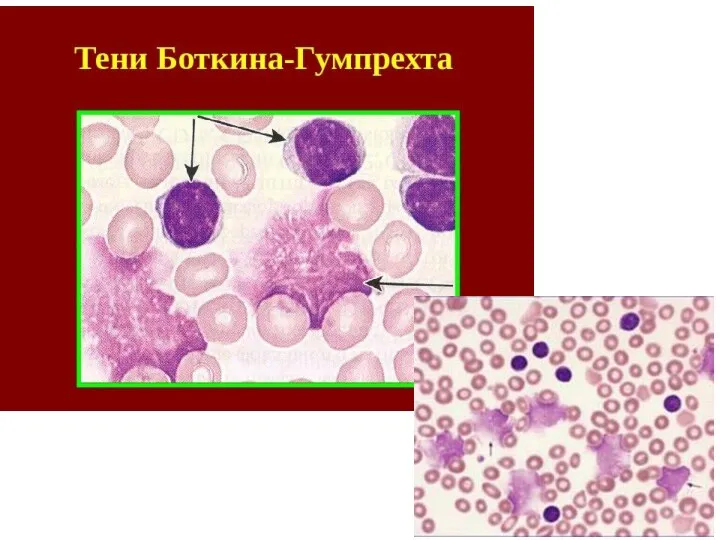

Волосатоклеточный лейкоз
Волосато-клеточный лейкоз – редкий вариант хронического лейкоза из хорошо дифференцированных
Волосатоклеточный лейкоз
Волосато-клеточный лейкоз – редкий вариант хронического лейкоза из хорошо дифференцированных

Диагностика
Костный мозг нормо- или гиперклеточный с диффузной лимфоидной инфильтрацией, часто развивается
Диагностика
Костный мозг нормо- или гиперклеточный с диффузной лимфоидной инфильтрацией, часто развивается
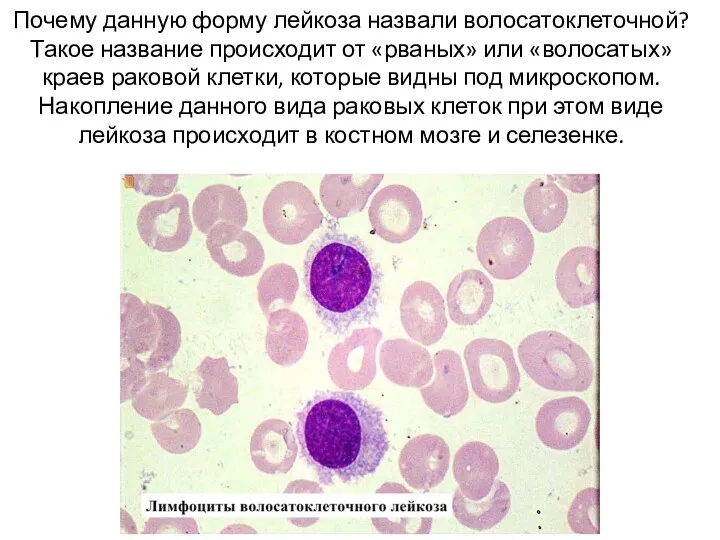
Почему данную форму лейкоза назвали волосатоклеточной? Такое название происходит от «рваных»
Почему данную форму лейкоза назвали волосатоклеточной? Такое название происходит от «рваных»

Парапротеинемические гемобластозы.
- опухоли системы В - лимфоцитов, дифференцирующиеся до стадии секреции
Парапротеинемические гемобластозы.
- опухоли системы В - лимфоцитов, дифференцирующиеся до стадии секреции
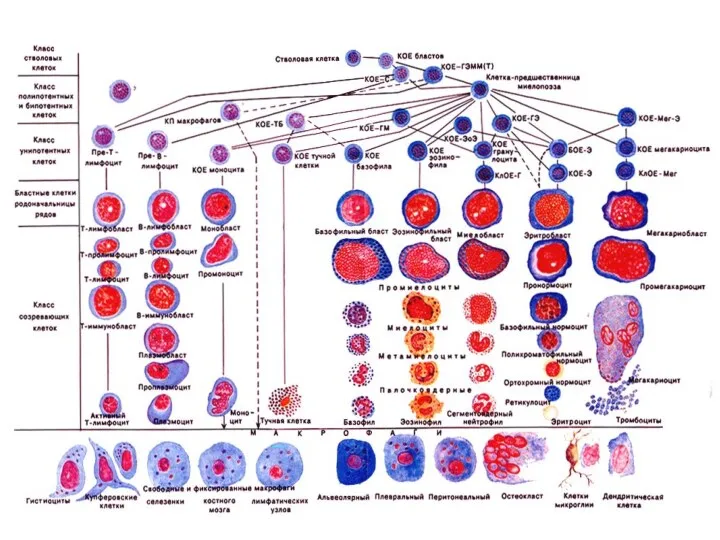
Схема кроветворения
Схема кроветворения

Парапротеинемические гемобластозы.
Патогенез.
Основной отличительной особенностью этих заболеваний является продукция моноклонального иммуноглобулина
Парапротеинемические гемобластозы.
Патогенез.
Основной отличительной особенностью этих заболеваний является продукция моноклонального иммуноглобулина
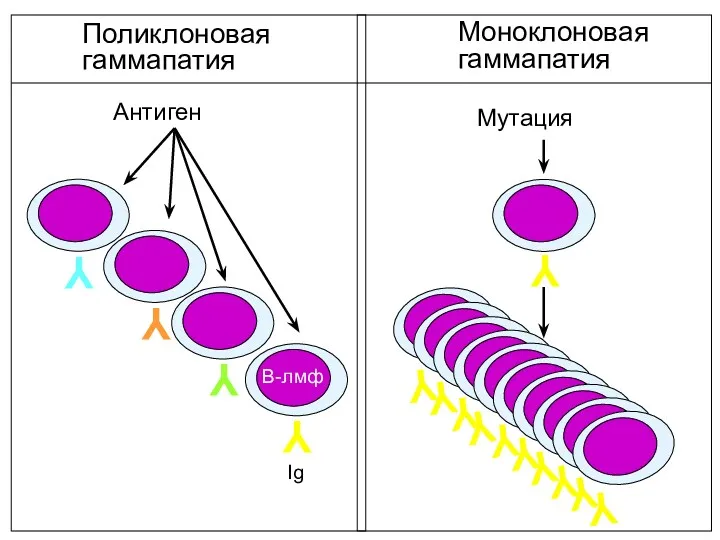
Y
Y
Y
Y
Антиген
Мутация
Y
Y
Y
Y
Y
Y
Y
Y
Y
Y
Y
Поликлоновая
гаммапатия
Моноклоновая
гаммапатия
Ig
В-лмф
Y
Y
Y
Y
Антиген
Мутация
Y
Y
Y
Y
Y
Y
Y
Y
Y
Y
Y
Поликлоновая
гаммапатия
Моноклоновая
гаммапатия
Ig
В-лмф

Парапротеинемические гемобластозы
Основную часть составляет IgG. При миеломе у большинства больных моноклональный
Парапротеинемические гемобластозы
Основную часть составляет IgG. При миеломе у большинства больных моноклональный

Лабораторная диагностика Парапротеинемических гемобластозов.
1) повышение СОЭ, которое является результатом увеличения
Лабораторная диагностика Парапротеинемических гемобластозов.
1) повышение СОЭ, которое является результатом увеличения

Лабораторная диагностика парапротеинов.
Разделение растворенных белков под действием постоянного электрического поля
Лабораторная диагностика парапротеинов.
Разделение растворенных белков под действием постоянного электрического поля
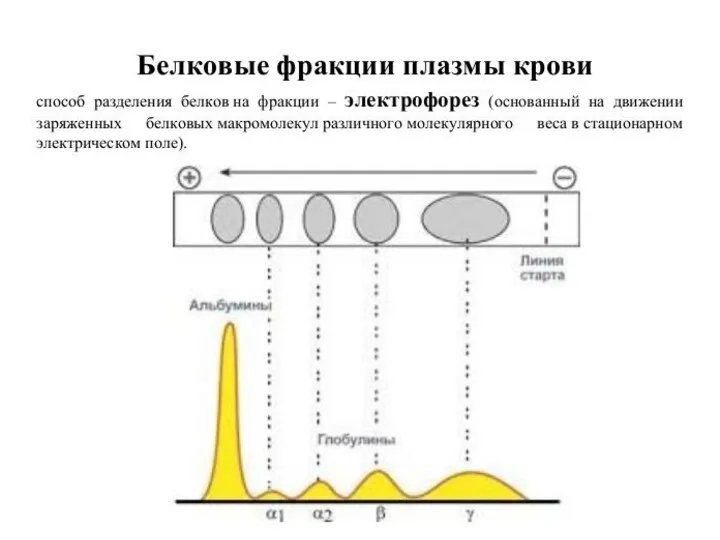
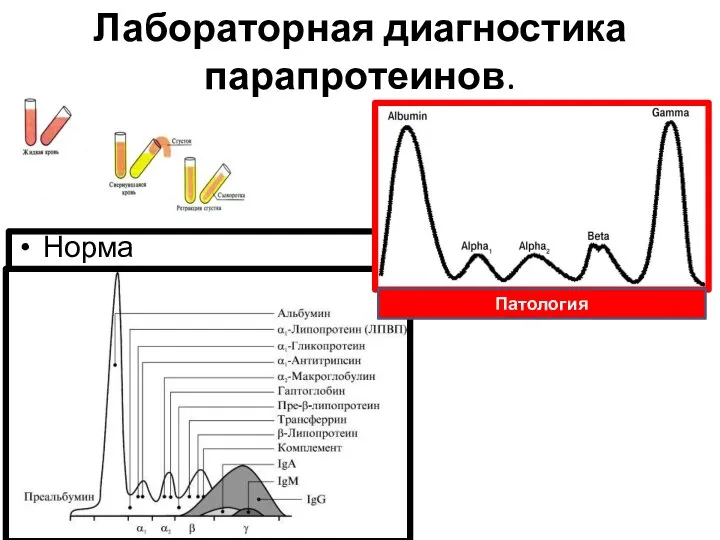
Лабораторная диагностика парапротеинов.
Норма
Патология
Лабораторная диагностика парапротеинов.
Норма
Патология
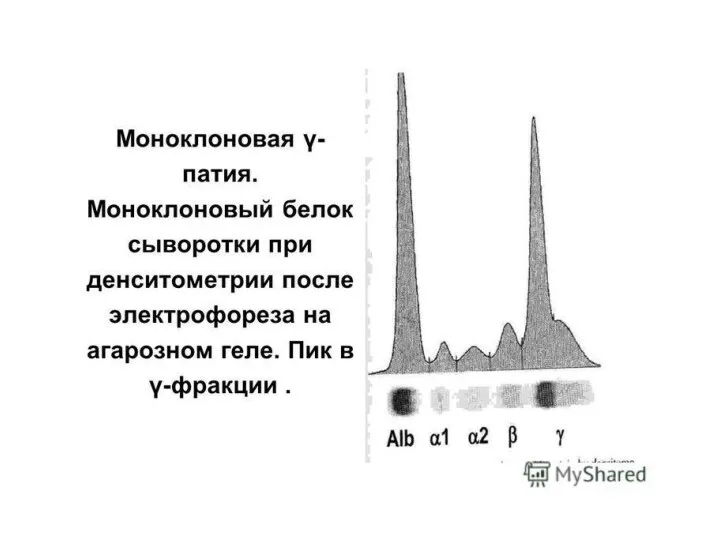

Парапротеинемические гемобластозы. Классификация.
Миеломная болезнь (плазмоцитома, множественная миелома ) — В-клеточное лимфопролиферативное заболевание,
Парапротеинемические гемобластозы. Классификация.
Миеломная болезнь (плазмоцитома, множественная миелома ) — В-клеточное лимфопролиферативное заболевание,

Миеломная болезнь (множественная миелома)
Миеломная болезнь — В-клеточное лимфопролиферативное заболевание, характеризующееся клональной
Миеломная болезнь (множественная миелома)
Миеломная болезнь — В-клеточное лимфопролиферативное заболевание, характеризующееся клональной

Этиология
1. Ионизирующая радиация
2. Генетическая предрасположенность к развитию МБ
3. Цитогенетические нарушения. Мутация
Этиология
1. Ионизирующая радиация
2. Генетическая предрасположенность к развитию МБ
3. Цитогенетические нарушения. Мутация

Иммунохимические варианты множественной миеломы
1. G-миелома 55-65%
2. А-миелома 20-25%
3. D-миелома 2-5%
4. Е-миелома
5.
1. G-миелома 55-65%
2. А-миелома 20-25%
3. D-миелома 2-5%
4. Е-миелома
5.
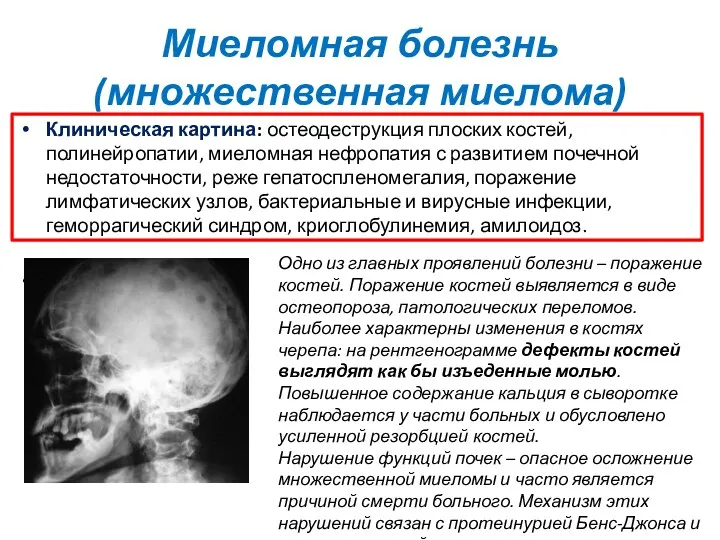
Миеломная болезнь (множественная миелома)
Клиническая картина: остеодеструкция плоских костей, полинейропатии, миеломная нефропатия
Миеломная болезнь (множественная миелома)
Клиническая картина: остеодеструкция плоских костей, полинейропатии, миеломная нефропатия

Диагностика
В костном мозге при миеломной болезни отмечается плазмоклеточная инфильтрация с
Диагностика
В костном мозге при миеломной болезни отмечается плазмоклеточная инфильтрация с
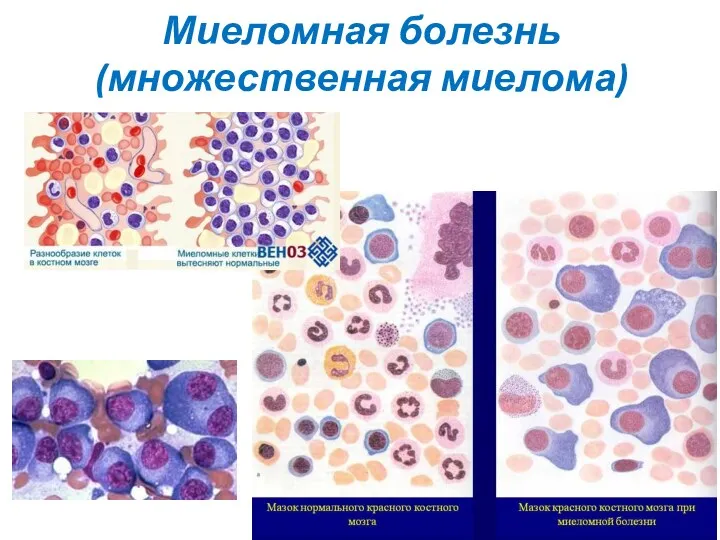
Миеломная болезнь (множественная миелома)
Миеломная болезнь (множественная миелома)
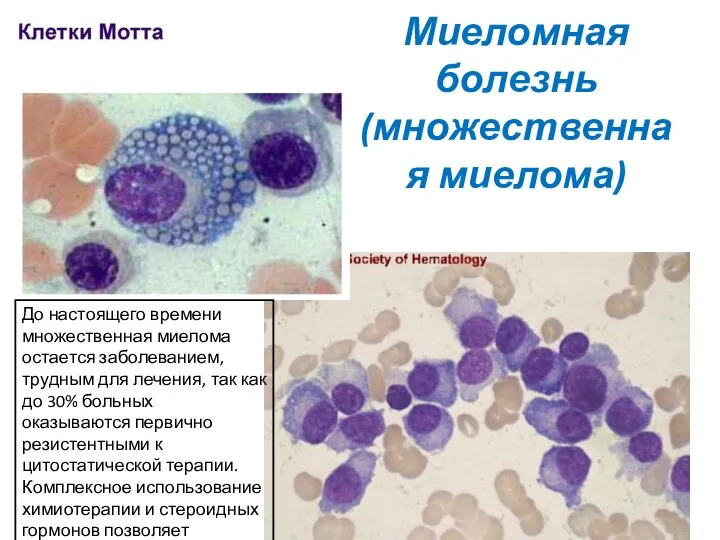
Миеломная болезнь (множественная миелома)
До настоящего времени множественная миелома остается заболеванием, трудным
Миеломная болезнь (множественная миелома)
До настоящего времени множественная миелома остается заболеванием, трудным

Макроглобулинемия Вальденстрема
Макроглобулинемия Вальденстрема — В-клеточная опухоль, морфологически представленная лимфоцитами, зрелыми плазматическими
Макроглобулинемия Вальденстрема
Макроглобулинемия Вальденстрема — В-клеточная опухоль, морфологически представленная лимфоцитами, зрелыми плазматическими

Макроглобулинемия Вальденстрема
Формы течения: бессимптомная, медленно прогрессирующая (продолжительность жизни более 5 лет),
Макроглобулинемия Вальденстрема
Формы течения: бессимптомная, медленно прогрессирующая (продолжительность жизни более 5 лет),

Клинические проявления МВ.
Обусловлены пролиферацией лимфоидных элементов в костном мозге, печени,
Клинические проявления МВ.
Обусловлены пролиферацией лимфоидных элементов в костном мозге, печени,

Макроглобулинемия Вальденстрема
Диагностика
В костном мозге отмечаются пролиферация лимфоцитов, иногда с плазматизированной цитоплазмой,
Макроглобулинемия Вальденстрема
Диагностика
В костном мозге отмечаются пролиферация лимфоцитов, иногда с плазматизированной цитоплазмой,
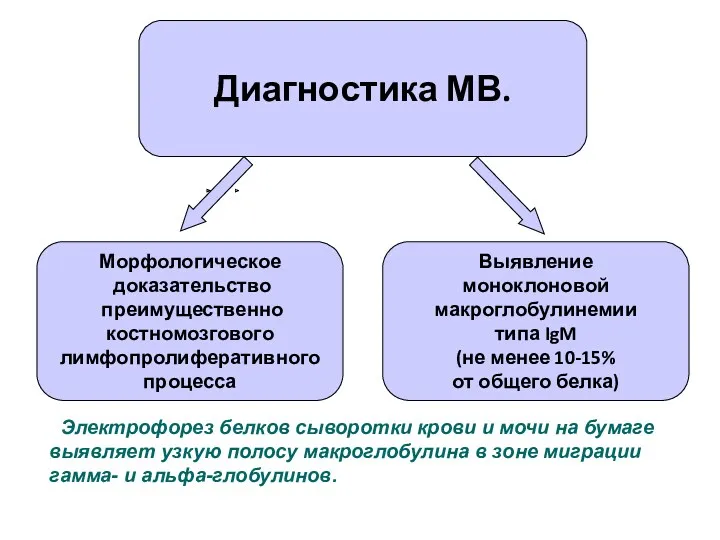
Морфологическое
доказательство
преимущественно
костномозгового
лимфопролиферативного
процесса
Выявление
моноклоновой
макроглобулинемии
типа IgM
(не менее 10-15%
от общего белка)
Диагностика МВ.
Электрофорез
Морфологическое
доказательство
преимущественно
костномозгового
лимфопролиферативного
процесса
Выявление
моноклоновой
макроглобулинемии
типа IgM
(не менее 10-15%
от общего белка)
Диагностика МВ.
Электрофорез

Лимфомы
Лимфома — группа гематологических заболеваний лимфатической ткани, характеризующихся увеличением лимфатических узлов и/или поражением различных
Лимфомы
Лимфома — группа гематологических заболеваний лимфатической ткани, характеризующихся увеличением лимфатических узлов и/или поражением различных

Лимфомы
Термином неходжкинские лимфомы обозначают довольно большую группу лимфом, которые не являются
Лимфомы
Термином неходжкинские лимфомы обозначают довольно большую группу лимфом, которые не являются
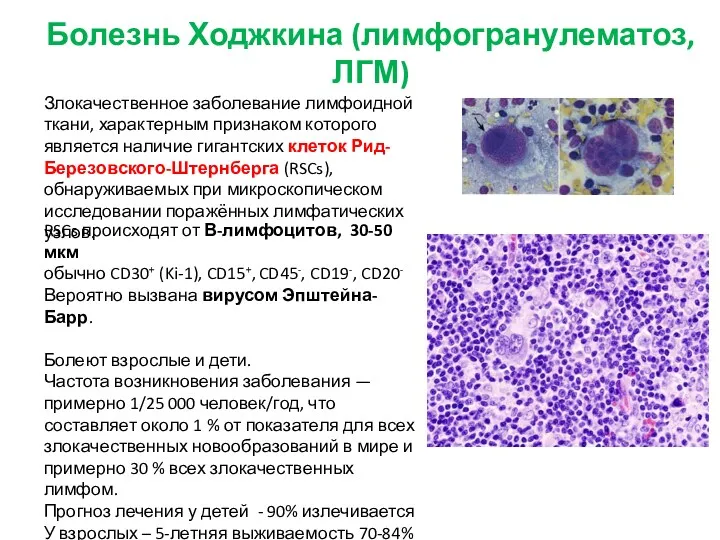
Болезнь Ходжкина (лимфогранулематоз, ЛГМ)
Злокачественное заболевание лимфоидной ткани, характерным признаком которого является
Болезнь Ходжкина (лимфогранулематоз, ЛГМ)
Злокачественное заболевание лимфоидной ткани, характерным признаком которого является

Болезнь Ходжкина (лимфогранулематоз, ЛГМ)
Патогенез. Опухолевым субстратом ЛГМ являются специфические гигантские клетки
Болезнь Ходжкина (лимфогранулематоз, ЛГМ)
Патогенез. Опухолевым субстратом ЛГМ являются специфические гигантские клетки

Неходжкинские лимфомы
Неходжкинские лимфомы являются гетерогенной группой неопластических заболеваний, происходящих из иммунной
Неходжкинские лимфомы
Неходжкинские лимфомы являются гетерогенной группой неопластических заболеваний, происходящих из иммунной

Этиология неходжкинских лимфом
Инфекционные
Вирус Эпштейна-Барр — ассоциирован с лимфомой Бёркитта, лимфогранулематозом, фолликулярной дендритно-клеточной саркомой,
Этиология неходжкинских лимфом
Инфекционные
Вирус Эпштейна-Барр — ассоциирован с лимфомой Бёркитта, лимфогранулематозом, фолликулярной дендритно-клеточной саркомой,

Неходжкинские лимфомы
Клиническая картина. Наиболее часто в дебюте заболевания появляется опухоль лимфатического
Неходжкинские лимфомы
Клиническая картина. Наиболее часто в дебюте заболевания появляется опухоль лимфатического

Неходжкинские лимфомы
Общие симптомы:
высокая температура (выше 38°C), причина её появления непонятна [симптом
Неходжкинские лимфомы
Общие симптомы:
высокая температура (выше 38°C), причина её появления непонятна [симптом

Неходжкинские лимфомы
Диагностика. Диагноз лимфомы основывается на исследовании морфологического субстрата опухоли. Обычно
Неходжкинские лимфомы
Диагностика. Диагноз лимфомы основывается на исследовании морфологического субстрата опухоли. Обычно

Неходжкинские лимфомы
Основной метод диагностики лимфомы – исследование поражённого лимфоузла (лимфатические узлы)
Неходжкинские лимфомы
Основной метод диагностики лимфомы – исследование поражённого лимфоузла (лимфатические узлы)

Стадирование лимфом
Стадирование лимфом

Неходжкинские лимфомы
Неходжкинские лимфомы
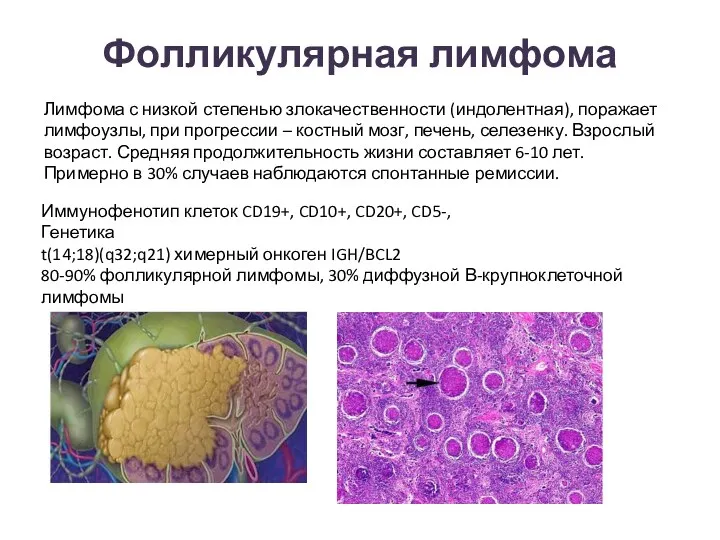
Фолликулярная лимфома
Иммунофенотип клеток CD19+, CD10+, CD20+, CD5-,
Генетика
t(14;18)(q32;q21) химерный онкоген IGH/BCL2
80-90%
Фолликулярная лимфома
Иммунофенотип клеток CD19+, CD10+, CD20+, CD5-,
Генетика
t(14;18)(q32;q21) химерный онкоген IGH/BCL2
80-90%

Фолликулярная лимфома
t(14;18)(q32;q21) химерный онкоген IGH/BCL2
80-90% фолликулярной лимфомы, 30% диффузной В-крупноклеточной лимфомы
Leukemia
Фолликулярная лимфома
t(14;18)(q32;q21) химерный онкоген IGH/BCL2
80-90% фолликулярной лимфомы, 30% диффузной В-крупноклеточной лимфомы
Leukemia
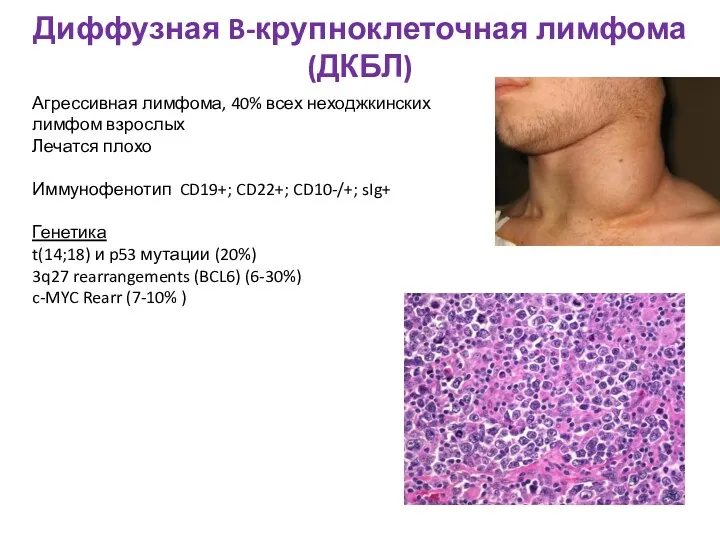
Диффузная B-крупноклеточная лимфома (ДКБЛ)
Агрессивная лимфома, 40% всех неходжкинских лимфом взрослых
Лечатся плохо
Иммунофенотип
Диффузная B-крупноклеточная лимфома (ДКБЛ)
Агрессивная лимфома, 40% всех неходжкинских лимфом взрослых
Лечатся плохо
Иммунофенотип
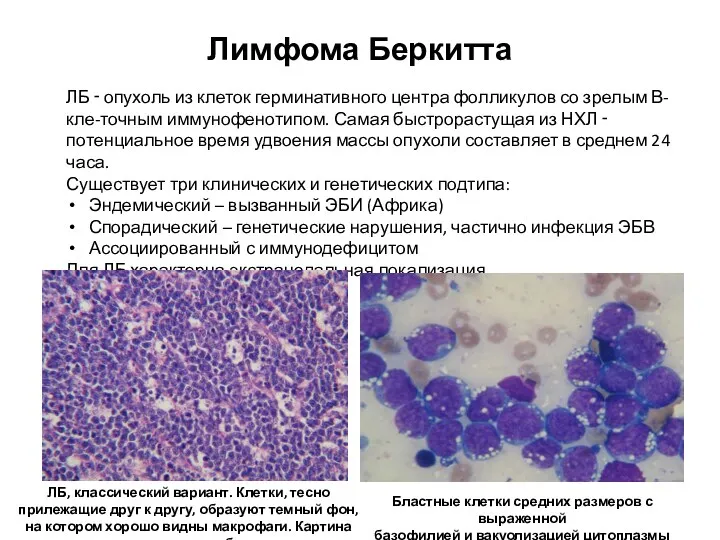
Лимфома Беркитта
ЛБ ‑ опухоль из клеток герминативного центра фолликулов со
Лимфома Беркитта
ЛБ ‑ опухоль из клеток герминативного центра фолликулов со
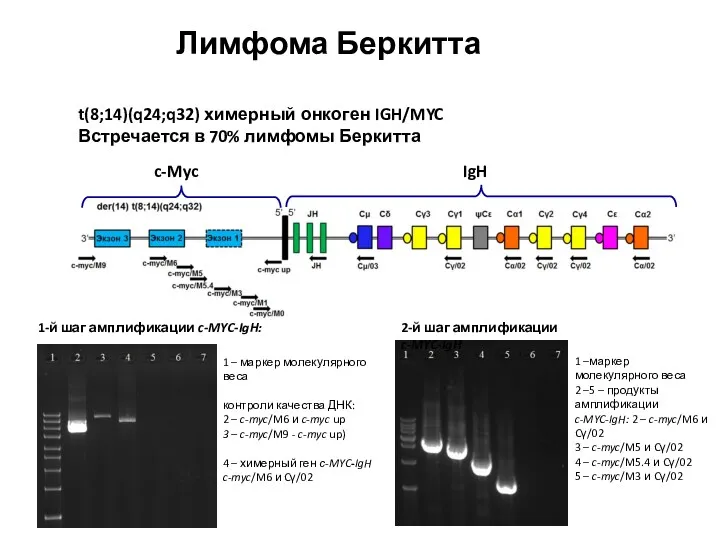
t(8;14)(q24;q32) химерный онкоген IGH/MYC
Встречается в 70% лимфомы Беркитта
c-Myc IgH
1 –
t(8;14)(q24;q32) химерный онкоген IGH/MYC
Встречается в 70% лимфомы Беркитта
c-Myc IgH
1 –

Грибовидный микоз/синдром Сезари (первичная Т-клеточная кожная лимфома)
Составляет 2-3% всех злокачественных лимфом.
Заболевание
Грибовидный микоз/синдром Сезари (первичная Т-клеточная кожная лимфома)
Составляет 2-3% всех злокачественных лимфом.
Заболевание
 Фитотерапия. Современная фитотерапия
Фитотерапия. Современная фитотерапия Ошибки и осложнения при лечении пострадавших с переломами костей и повреждениями суставов
Ошибки и осложнения при лечении пострадавших с переломами костей и повреждениями суставов Зат алмасу ауруларын тағаммен емдеу және емдік дене шынықтыру
Зат алмасу ауруларын тағаммен емдеу және емдік дене шынықтыру Долікарська допомога
Долікарська допомога Парентералды жолмен берілетін вирусты аурулар (ЖИТС) және олардың алдын алу шаралары
Парентералды жолмен берілетін вирусты аурулар (ЖИТС) және олардың алдын алу шаралары Логопедическое заключение. Направления работы при различных речевых нарушениях
Логопедическое заключение. Направления работы при различных речевых нарушениях Заикание. Этиология
Заикание. Этиология Сердечно- лёгочно-церебральная реанимация. Внебольничная остановка кровообращения
Сердечно- лёгочно-церебральная реанимация. Внебольничная остановка кровообращения Правила безопасного сексуального поведения
Правила безопасного сексуального поведения Медицинская генетика
Медицинская генетика Эпидемиология сахарного диабета (в РФ, субъектах РФ и других странах)
Эпидемиология сахарного диабета (в РФ, субъектах РФ и других странах) Бел, сегізкөз және құйымшақ жұлын нервтері
Бел, сегізкөз және құйымшақ жұлын нервтері Bronchitis in children
Bronchitis in children Тырыспаға қарсы және психотроптық дәрілік заттардың клиникалық фармакологиясы
Тырыспаға қарсы және психотроптық дәрілік заттардың клиникалық фармакологиясы Хронофармакология
Хронофармакология Заболевания сосудов. Нарушения венозного оттока
Заболевания сосудов. Нарушения венозного оттока Организация доклинических испытаний лекарственных препаратов
Организация доклинических испытаний лекарственных препаратов Қазіргі карпульды анестетиктер. Түрлері. Клиникалық фармокологиялық сипаттамасы. Стоматологиядағы жергілікті инъекциялық
Қазіргі карпульды анестетиктер. Түрлері. Клиникалық фармокологиялық сипаттамасы. Стоматологиядағы жергілікті инъекциялық Биологическая опасность. Понятие инфекционного и эпидемического процесса. Дезинфекция, дезинсекция. дератизация: понятия и виды
Биологическая опасность. Понятие инфекционного и эпидемического процесса. Дезинфекция, дезинсекция. дератизация: понятия и виды Анестезия в сердечно-сосудистой хирургии
Анестезия в сердечно-сосудистой хирургии Пиелонефрит у беременных
Пиелонефрит у беременных Переход к системе непрерывного медицинского (фармацевтического) образования
Переход к системе непрерывного медицинского (фармацевтического) образования Врачебные специальности. Послевузовсое образование врача-лечебника
Врачебные специальности. Послевузовсое образование врача-лечебника Психопатии. Патохарактерологическое развитие личности
Психопатии. Патохарактерологическое развитие личности Cтоп коронавирус
Cтоп коронавирус Панариций. Классификация и причины панариций
Панариций. Классификация и причины панариций Медицинада ДНҚ-диагностика әдісін қолдану
Медицинада ДНҚ-диагностика әдісін қолдану Фармакотерапия при гипертонической болезни
Фармакотерапия при гипертонической болезни