Мінливість та її види. Зчеплене успадкування. Методи вивчення генетики людини. (Лекция 6) презентация
- Мінливість та її види. Зчеплене успадкування. Методи вивчення генетики людини. (Лекция 6)

Содержание
- 2. Мінливість: її причини та методи вивчення 1.Класифікація мінливості. 2.Спадкова мінливість: Генні мутації Хромосомні мутації Геномні мутації
- 3. Мінливість Спадкова мінливість – це здатність до зміни генетичного матеріалу. Поділяється на : комбінативну та мутаційну.
- 4. Модифікаційна мінливість
- 5. Основні положення мутаційної теорії Г. Де Фріза (1901 – 1903) Мутації виникають несподівано як дискретні зміни
- 6. Мутації та модифікації
- 7. Класифікація мутацій 1. За характером зміни генома: Геномні мутації – зміни числа хромосом. Хромосомні мутації –
- 8. Класифікація мутацій 5. За локалізацією в клітині: Ядерні Цитоплазматичні 6. За відношенням до можливості успадкування: Генеративні,
- 9. Гені мутації Основна увага при вивченні генних мутацій приділяється змінам чергування пар нуклеотидів в ДНК і
- 10. Генні мутації
- 11. Генні мутації
- 12. Хромосомні мутації 1. Внутріхромосомні перебудови: Дефішенсі – втрата кінцьової ділянки хромосоми Делеції – випадіння частини хромосоми,
- 13. Хромосомні мутації
- 14. Геномні мутації Анеуплоїдія – це зміна числа хромосом, яка не кратна гаплоїдному набору хромосом. Поліплоїдія –
- 15. Види анеуплоїдів і поліплоїдів
- 16. Типи аутополіплоїдизації
- 17. Анеуплоїдія
- 18. Успадкування генів, які містяться в одній хромосомі
- 19. Успадкування генів, які містяться в одній хромосомі
- 20. Кросинговер
- 21. Хромосомна теорія спадковості Основні положення хромосомної теорії спадковості: Гени локалізовані в хромосомах. Кожна хромосома – це
- 22. Генетичні карти хромосом
- 23. Генеалогічний метод
- 24. Генеалогічний метод
- 25. Успадкування ознак які мають просту (моногенну) природу Домінантна ознака передається без пропусків поколінь. Рецесивна ознака передається
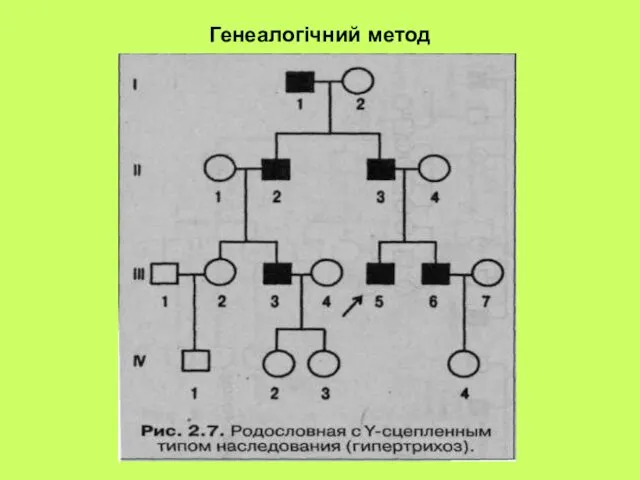
- 26. Генеалогічний метод
- 27. Генеалогічний метод
- 29. Генеалогічний метод
- 30. Близнюковий метод
- 31. Близнюковий метод Близнюковий метод – метод оцінки співвідносної ролі спадковості і середовища у становленні фенотипу, заснований
- 32. Близнюковий метод використовується у генетиці людини для того, щоб оцінити ступінь впливу спадковості і середовища на
- 33. Близнюковий метод
- 34. Популяційно-статистичний метод За допомогою цього методу вивчають генетичну структуру популяцій, їх генофонд, взаємодію факторів, які обумовлюють
- 35. Популяційно-статистичний метод Досліджувані популяції можуть розрізнятися за біологічними ознаками, географічними умовами життя, економічним станом. Категорії генів
- 36. Популяційно-статистичний метод 1908 рік – закон Харді-Вайнберга: в ідеальній популяції співвідношення частоти домінантних гомозигот (АА), гетерозигот
- 37. Цитогенетичний метод Заснований на мікроскопічному вивченні хромосом. Метод дозволяє: вивчати стандартний каріотип людини; виявити спадкові хвороби,
- 38. Біохімічні методи При різних типах захворювання вдається або визначити сам аномальний білок — фермент, або проміжні
- 39. Спадкові хвороби людини Генні хвороби пов’язані із змінами в структурі генів, передаються від покоління до покоління
- 40. Мутації Генні мутації Велика частина генних мутацій призводить до синтезу дефектного білка, який не здатний виконувати
- 41. Класифікація генних спадкових хвороб за характером метаболічних розладів – порушення обміну амінокислот (приклади: фенілпіро- виноградна олігофренія,
- 42. Генні хвороби Ланцюг послідовності формування генних захворювань В результаті мутацій гена на молекулярному рівні можливі наступні
- 43. Генні хвороби Загальна частота генних хвороб популяції складає 1-2%. Моногенні форми генних хвороб успадковуються за законами
- 44. ХРОМОСОМНІ ХВОРОБИ (ВІДОМО ПОНАД 700 ЗАХВОРЮВАНЬ) Виникають в результаті мутацій хромосом статевих клітин одного з батьків
- 45. Спадкові хвороби, викликані нерозходженням аутосом Нерозходження від 1 до 12 пар хромосом викликає смерть. - Нерозходження
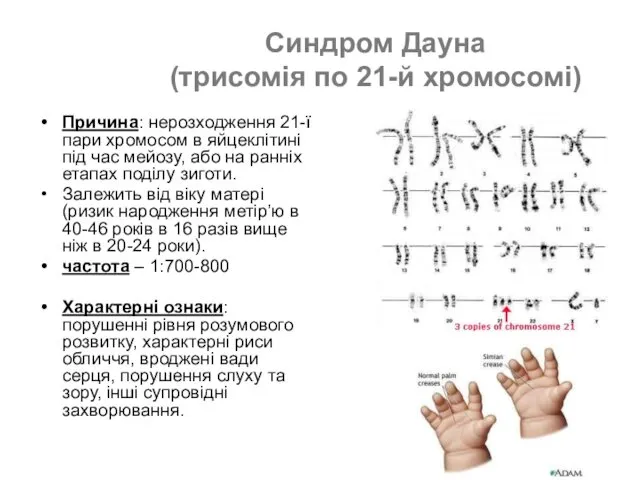
- 46. Синдром Дауна (трисомія по 21-й хромосомі) Причина: нерозходження 21-ї пари хромосом в яйцеклітині під час мейозу,
- 47. Люди з синдром Дауна (трисомія по 21-й хромосомі)
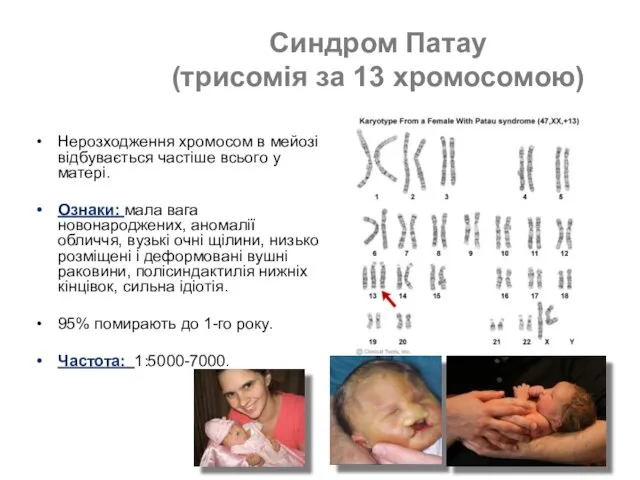
- 48. Синдром Патау (трисомія за 13 хромосомою) Нерозходження хромосом в мейозі відбувається частіше всього у матері. Ознаки:
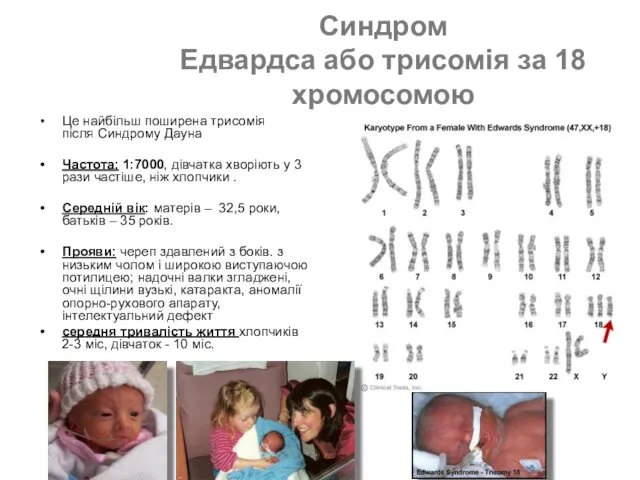
- 49. Синдром Едвардса або трисомія за 18 хромосомою Це найбільш поширена трисомія після Синдрому Дауна Частота: 1:7000,
- 50. Синдром котячого крику (відомий під назвами: синдром делеції короткого плеча 5 хромосоми, 5р синдром або синдром
- 51. Фактори підвищення ризику народження дітей з хромосомними захворюваннями Іонізуюче випромінювання; віруси; Хімічні речовини; Вік батьків; Прийом
- 52. МЕДИКО-ГЕНЕТИЧНЕ КОНСУЛЬТУВАННЯ Під час консультації лікар має допомогти пацієнту/родині: проаналізувати медичну інформацію, включаючи встановлений діагноз, можливий
- 53. Покази до проведення преконцепційної та пренатальної медико-генетичної консультації: Зрілий вік матері (35 та більше років під
- 54. Основні методи пренатальної діагностики Визначення альфа-фетопротеїну Ультразвукове дослідження плоду Біопсія хоріону та плаценти Амніоцентез Кордоцентез Фетоскопія
- 55. Пренатальна діагностика
- 56. МУЛЬТИФАКТОРІАЛЬНІ ХВОРОБИ 92% від всіх спадкових патологій Найбільш поширені хвороби Ревматизм; Ішемічна хвороба; Гіпертонічна хвороба; Виразкова
- 58. Скачать презентацию

Мінливість: її причини та методи вивчення
1.Класифікація мінливості.
2.Спадкова мінливість:
Генні мутації
Хромосомні мутації
Геномні мутації
3.
Мінливість: її причини та методи вивчення
1.Класифікація мінливості.
2.Спадкова мінливість:
Генні мутації
Хромосомні мутації
Геномні мутації
3.

Мінливість
Спадкова мінливість – це здатність до зміни генетичного матеріалу. Поділяється на
Мінливість
Спадкова мінливість – це здатність до зміни генетичного матеріалу. Поділяється на

Модифікаційна мінливість
Модифікаційна мінливість

Основні положення мутаційної теорії
Г. Де Фріза (1901 – 1903)
Мутації виникають
Основні положення мутаційної теорії
Г. Де Фріза (1901 – 1903)
Мутації виникають

Мутації та модифікації
Мутації та модифікації

Класифікація мутацій
1. За характером зміни генома:
Геномні мутації – зміни числа хромосом.
Хромосомні
Класифікація мутацій
1. За характером зміни генома:
Геномні мутації – зміни числа хромосом.
Хромосомні

Класифікація мутацій
5. За локалізацією в клітині:
Ядерні
Цитоплазматичні
6. За відношенням до можливості успадкування:
Генеративні,
Класифікація мутацій
5. За локалізацією в клітині:
Ядерні
Цитоплазматичні
6. За відношенням до можливості успадкування:
Генеративні,

Гені мутації
Основна увага при вивченні генних мутацій приділяється змінам чергування пар
Гені мутації
Основна увага при вивченні генних мутацій приділяється змінам чергування пар

Генні мутації
Генні мутації
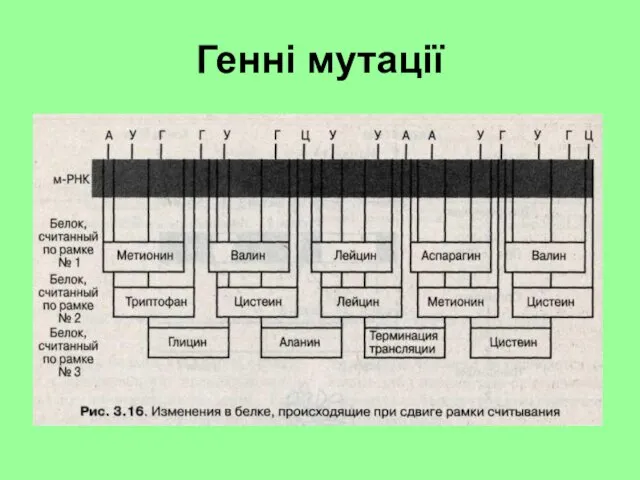
Генні мутації
Генні мутації

Хромосомні мутації
1. Внутріхромосомні перебудови:
Дефішенсі – втрата кінцьової ділянки хромосоми
Делеції – випадіння
Хромосомні мутації
1. Внутріхромосомні перебудови:
Дефішенсі – втрата кінцьової ділянки хромосоми
Делеції – випадіння
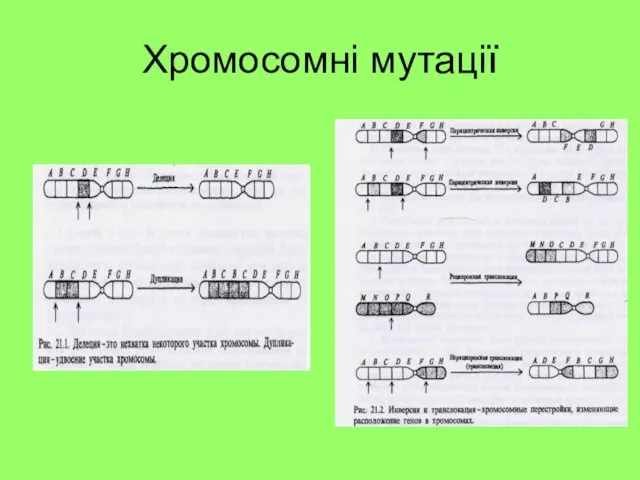
Хромосомні мутації
Хромосомні мутації

Геномні мутації
Анеуплоїдія – це зміна числа хромосом, яка не кратна гаплоїдному
Геномні мутації
Анеуплоїдія – це зміна числа хромосом, яка не кратна гаплоїдному
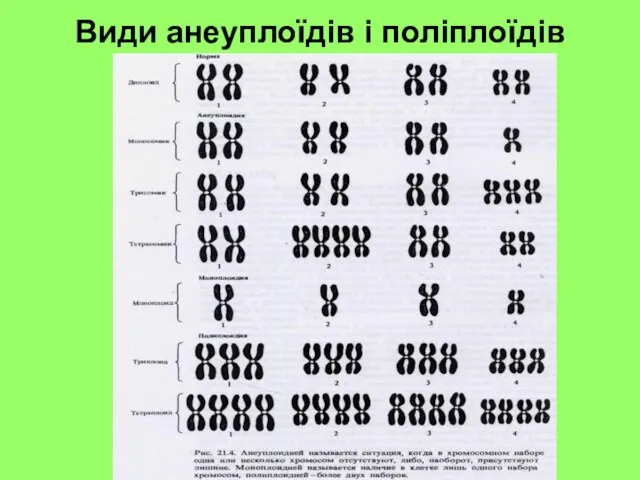
Види анеуплоїдів і поліплоїдів
Види анеуплоїдів і поліплоїдів
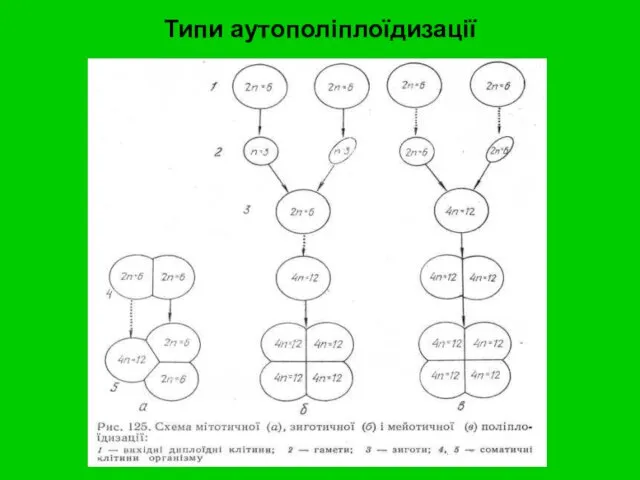
Типи аутополіплоїдизації
Типи аутополіплоїдизації
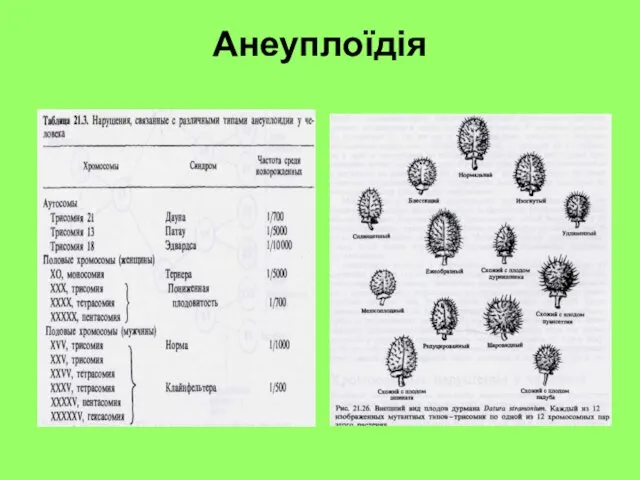
Анеуплоїдія
Анеуплоїдія

Успадкування генів, які містяться в одній хромосомі
Успадкування генів, які містяться в одній хромосомі
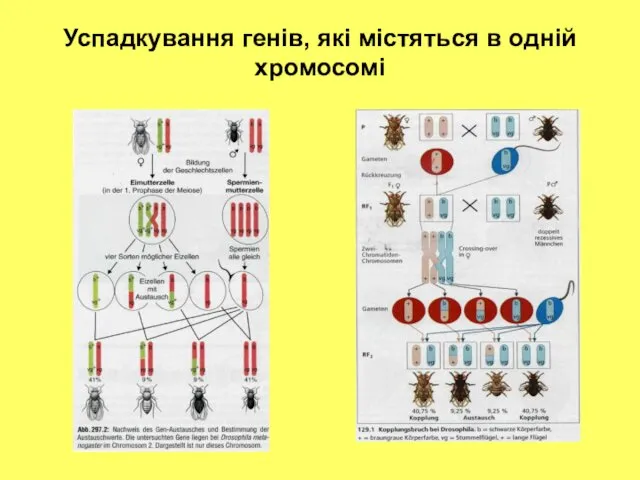
Успадкування генів, які містяться в одній хромосомі
Успадкування генів, які містяться в одній хромосомі

Кросинговер
Кросинговер

Хромосомна теорія спадковості
Основні положення хромосомної теорії спадковості:
Гени локалізовані в хромосомах. Кожна
Хромосомна теорія спадковості
Основні положення хромосомної теорії спадковості:
Гени локалізовані в хромосомах. Кожна
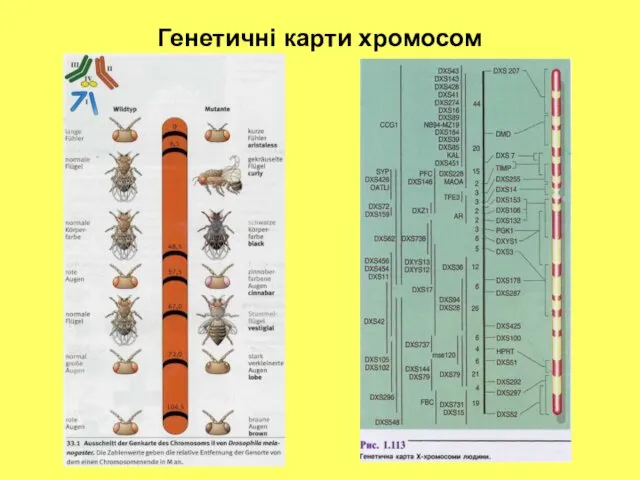
Генетичні карти хромосом
Генетичні карти хромосом

Генеалогічний метод
Генеалогічний метод

Генеалогічний метод
Генеалогічний метод

Успадкування ознак які мають просту (моногенну) природу
Домінантна ознака передається без пропусків
Успадкування ознак які мають просту (моногенну) природу
Домінантна ознака передається без пропусків
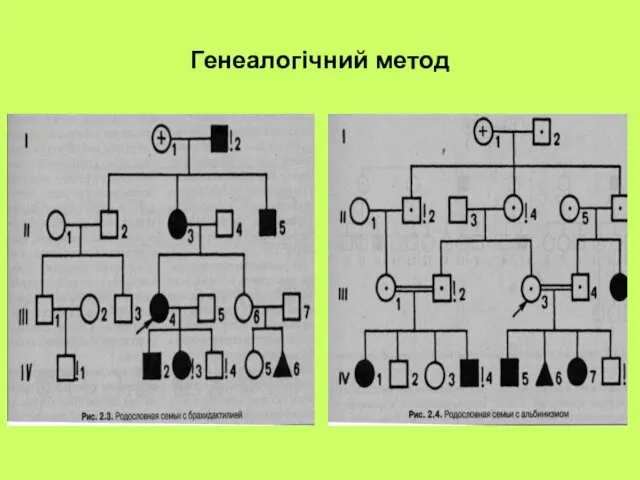
Генеалогічний метод
Генеалогічний метод
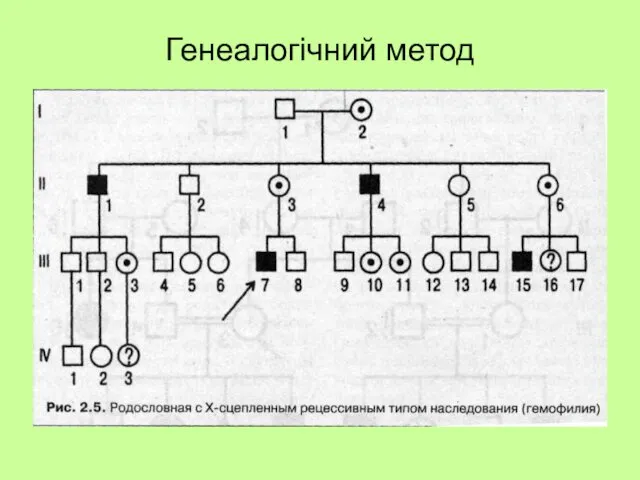
Генеалогічний метод
Генеалогічний метод
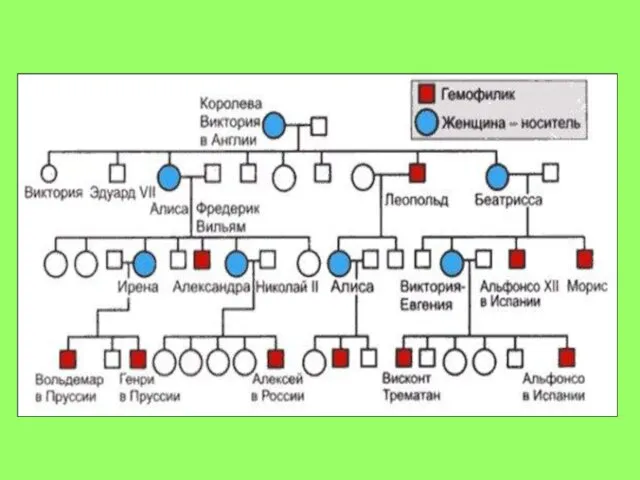
Генеалогічний метод
Генеалогічний метод

Близнюковий метод
Близнюковий метод

Близнюковий метод
Близнюковий метод – метод оцінки співвідносної ролі спадковості і
Близнюковий метод
Близнюковий метод – метод оцінки співвідносної ролі спадковості і

Близнюковий метод використовується у генетиці людини для того, щоб оцінити ступінь
Близнюковий метод використовується у генетиці людини для того, щоб оцінити ступінь
Близнюковий метод
Близнюковий метод

Популяційно-статистичний метод
За допомогою цього методу вивчають генетичну структуру популяцій, їх генофонд,
Популяційно-статистичний метод
За допомогою цього методу вивчають генетичну структуру популяцій, їх генофонд,

Популяційно-статистичний метод
Досліджувані популяції можуть розрізнятися за біологічними ознаками, географічними умовами
Популяційно-статистичний метод
Досліджувані популяції можуть розрізнятися за біологічними ознаками, географічними умовами

Популяційно-статистичний метод
1908 рік – закон Харді-Вайнберга: в ідеальній популяції співвідношення частоти
Популяційно-статистичний метод
1908 рік – закон Харді-Вайнберга: в ідеальній популяції співвідношення частоти
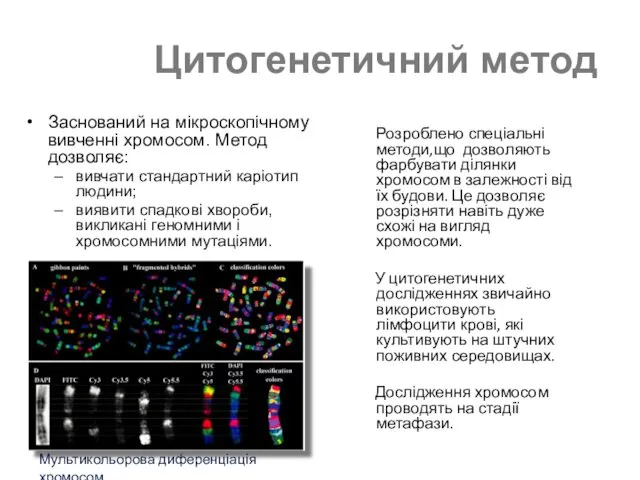
Цитогенетичний метод
Заснований на мікроскопічному вивченні хромосом. Метод дозволяє:
вивчати стандартний каріотип людини;
виявити
Цитогенетичний метод
Заснований на мікроскопічному вивченні хромосом. Метод дозволяє:
вивчати стандартний каріотип людини;
виявити

Біохімічні методи
При різних типах захворювання вдається або визначити сам аномальний
Біохімічні методи
При різних типах захворювання вдається або визначити сам аномальний

Спадкові хвороби людини
Генні хвороби пов’язані із змінами в структурі генів, передаються
Спадкові хвороби людини
Генні хвороби пов’язані із змінами в структурі генів, передаються

Мутації
Генні мутації
Велика частина генних мутацій призводить до синтезу дефектного
Мутації
Генні мутації
Велика частина генних мутацій призводить до синтезу дефектного

Класифікація генних спадкових хвороб за характером метаболічних розладів
– порушення обміну амінокислот
Класифікація генних спадкових хвороб за характером метаболічних розладів
– порушення обміну амінокислот
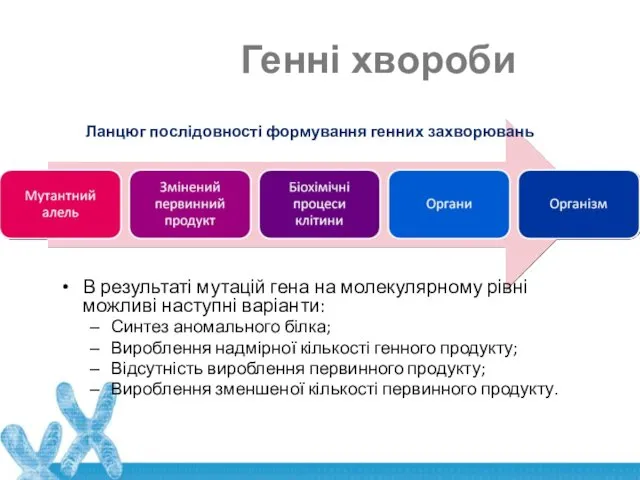
Генні хвороби
Ланцюг послідовності формування генних захворювань
В результаті мутацій гена на
Генні хвороби
Ланцюг послідовності формування генних захворювань
В результаті мутацій гена на
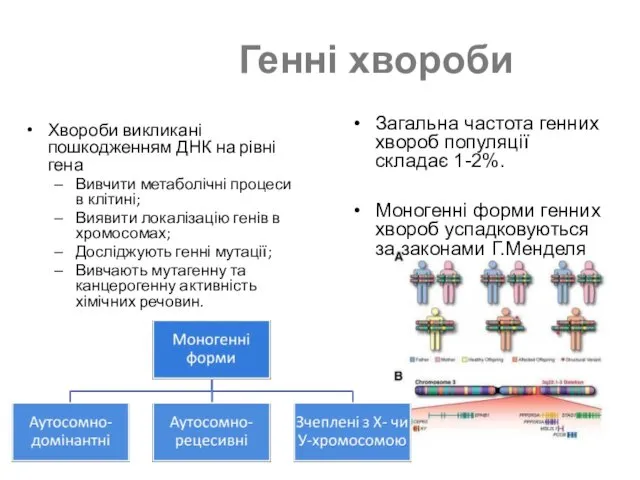
Генні хвороби
Загальна частота генних хвороб популяції складає 1-2%.
Моногенні форми генних
Генні хвороби
Загальна частота генних хвороб популяції складає 1-2%.
Моногенні форми генних
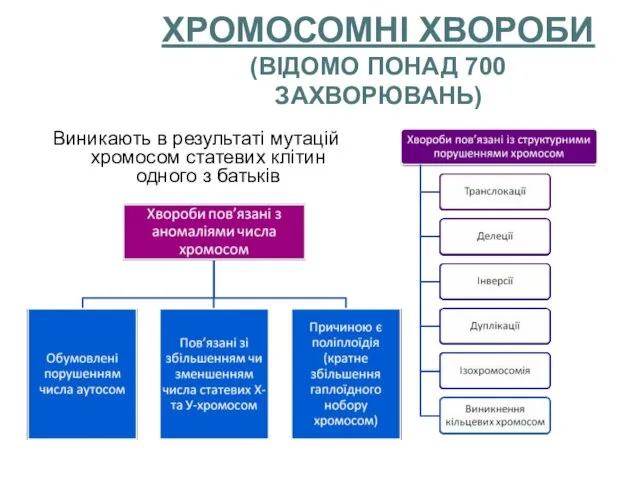
ХРОМОСОМНІ ХВОРОБИ
(ВІДОМО ПОНАД 700 ЗАХВОРЮВАНЬ)
Виникають в результаті мутацій хромосом статевих клітин
ХРОМОСОМНІ ХВОРОБИ
(ВІДОМО ПОНАД 700 ЗАХВОРЮВАНЬ)
Виникають в результаті мутацій хромосом статевих клітин

Спадкові хвороби, викликані нерозходженням аутосом
Нерозходження від 1 до 12
Спадкові хвороби, викликані нерозходженням аутосом
Нерозходження від 1 до 12
Синдром Дауна
(трисомія по 21-й хромосомі)
Причина: нерозходження 21-ї пари хромосом в
Синдром Дауна
(трисомія по 21-й хромосомі)
Причина: нерозходження 21-ї пари хромосом в

Люди з синдром Дауна
(трисомія по 21-й хромосомі)
Люди з синдром Дауна
(трисомія по 21-й хромосомі)
Синдром Патау
(трисомія за 13 хромосомою)
Нерозходження хромосом в мейозі відбувається частіше всього
Синдром Патау
(трисомія за 13 хромосомою)
Нерозходження хромосом в мейозі відбувається частіше всього
Синдром Едвардса або трисомія за 18 хромосомою
Це найбільш поширена трисомія після Синдрому Дауна
Частота: 1:7000,
Синдром Едвардса або трисомія за 18 хромосомою
Це найбільш поширена трисомія після Синдрому Дауна
Частота: 1:7000,
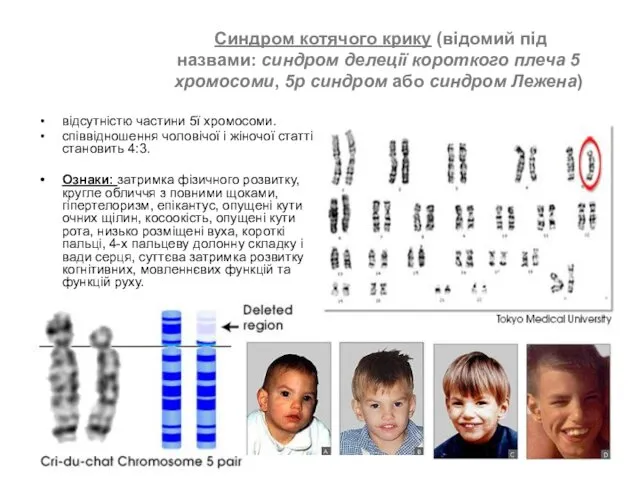
Синдром котячого крику (відомий під назвами: синдром делеції короткого плеча 5 хромосоми, 5р синдром або синдром Лежена)
відсутністю
Синдром котячого крику (відомий під назвами: синдром делеції короткого плеча 5 хромосоми, 5р синдром або синдром Лежена)
відсутністю

Фактори підвищення ризику народження дітей з хромосомними захворюваннями
Іонізуюче випромінювання;
віруси;
Хімічні речовини;
Вік батьків;
Прийом
Фактори підвищення ризику народження дітей з хромосомними захворюваннями
Іонізуюче випромінювання;
віруси;
Хімічні речовини;
Вік батьків;
Прийом

МЕДИКО-ГЕНЕТИЧНЕ КОНСУЛЬТУВАННЯ
Під час консультації лікар має допомогти пацієнту/родині:
проаналізувати медичну інформацію, включаючи
МЕДИКО-ГЕНЕТИЧНЕ КОНСУЛЬТУВАННЯ
Під час консультації лікар має допомогти пацієнту/родині:
проаналізувати медичну інформацію, включаючи

Покази до проведення преконцепційної та пренатальної медико-генетичної консультації:
Зрілий вік матері (35
Покази до проведення преконцепційної та пренатальної медико-генетичної консультації:
Зрілий вік матері (35

Основні методи пренатальної діагностики
Визначення альфа-фетопротеїну
Ультразвукове дослідження плоду
Біопсія хоріону та плаценти
Амніоцентез
Кордоцентез
Фетоскопія
Основні методи пренатальної діагностики
Визначення альфа-фетопротеїну
Ультразвукове дослідження плоду
Біопсія хоріону та плаценти
Амніоцентез
Кордоцентез
Фетоскопія

Пренатальна діагностика
Пренатальна діагностика
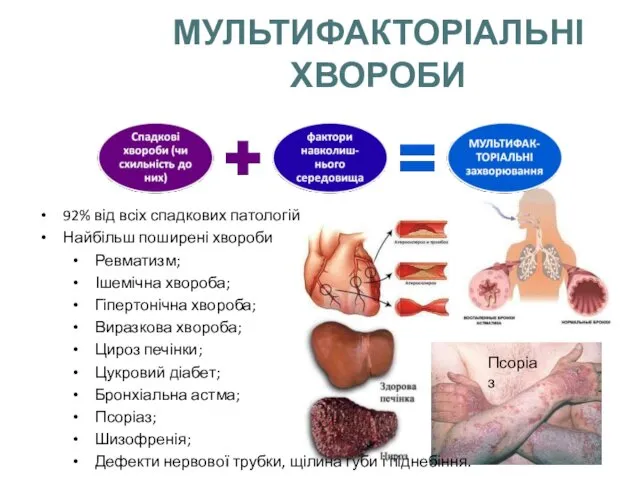
МУЛЬТИФАКТОРІАЛЬНІ ХВОРОБИ
92% від всіх спадкових патологій
Найбільш поширені хвороби
Ревматизм;
Ішемічна хвороба;
Гіпертонічна хвороба;
Виразкова
МУЛЬТИФАКТОРІАЛЬНІ ХВОРОБИ
92% від всіх спадкових патологій
Найбільш поширені хвороби
Ревматизм;
Ішемічна хвороба;
Гіпертонічна хвороба;
Виразкова
 Специфика инхаус в фармацевтической компании
Специфика инхаус в фармацевтической компании Эвтаназия
Эвтаназия Острые воспалительные заболевания органов малого таза
Острые воспалительные заболевания органов малого таза Роль среднего медицинского персонала, в профилактике кишечных инфекций
Роль среднего медицинского персонала, в профилактике кишечных инфекций Кровоснабжение головного мозга
Кровоснабжение головного мозга История неврологии
История неврологии Особенности сестринского обследования и ухода, при остром лейкозе у детей
Особенности сестринского обследования и ухода, при остром лейкозе у детей Постинъекционные осложнения. Роль средних медицинских работников в профилактике и лечении данной патологии
Постинъекционные осложнения. Роль средних медицинских работников в профилактике и лечении данной патологии Заболевания щитовидной железы и беременность
Заболевания щитовидной железы и беременность Митохондриальные болезни или опять во всем виноваты женщины
Митохондриальные болезни или опять во всем виноваты женщины Неврологиялық науқастарды реабилитациялау негіздері
Неврологиялық науқастарды реабилитациялау негіздері Дистрофии. Паранхиматозные дистрофии
Дистрофии. Паранхиматозные дистрофии Оригинальные препараты. Бренды и дженерики
Оригинальные препараты. Бренды и дженерики Острые лейкозы у детей
Острые лейкозы у детей Здоровье лиц пожилого и старческого возраста
Здоровье лиц пожилого и старческого возраста Речь и ее развитие в онтогенезе
Речь и ее развитие в онтогенезе Сүйек сыну. Буын шығу соғып алу. Сіңір созылу
Сүйек сыну. Буын шығу соғып алу. Сіңір созылу Үйреншікті жүктілікті тастау. Жүктіліктен тыс тексеріс жүктілікті жүргізу
Үйреншікті жүктілікті тастау. Жүктіліктен тыс тексеріс жүктілікті жүргізу Балалардағы бронх демікпесі
Балалардағы бронх демікпесі Зондирование
Зондирование Адренергические средства
Адренергические средства Личностные особенности пациентов с различными хроническими соматическими заболеваниями
Личностные особенности пациентов с различными хроническими соматическими заболеваниями Алкоголизм, алкогольные психозы
Алкоголизм, алкогольные психозы Внутриутробное развитие организма. Развитие после рождения
Внутриутробное развитие организма. Развитие после рождения Complicated cataract
Complicated cataract АИВ-инфекция және жүктілік
АИВ-инфекция және жүктілік Реєстрація лікарських засобів. Фармацевтична розробка
Реєстрація лікарських засобів. Фармацевтична розробка Учение об инфекции. Патогенность и вирулентность микробов
Учение об инфекции. Патогенность и вирулентность микробов