- Моногенные болезни

Содержание
- 2. Основные свойства моногенных болезней 1)Менделирующий характер наследования 2)Хронический крогра 3)Генетическая гетерогенность 4) Клинический полиморфизм Основным признаком,
- 3. Моногенные болезни можно разделить по типу наследования 1)Аутосомно-доминантные 2)Аутосомно-рецессивные 3)Х - Сцепленные доминантные 4)Х – Сцепленные
- 4. Никакие другие группы наследственной патологии (хромосомные и мультифакториальные болезни) таким образом не наследуются. Другим свойством моногенных
- 5. Моногенные болезни это заболевания, протекающие с постоянным прогрессированием патологического процесса.
- 6. Генетическая гетерогенность заключается в том, что развитие сходного фенотипа (то есть клинической картины) может быть обусловлено
- 7. Генетическую гетерогенность можно рассмотреть на примере врожденного гипотиреоза.Недостаточность продукции гормонов щитовидной железы при этом заболевании в
- 8. 1.Поступление неорганического йода в организм снижено из-за недостатка его в пище и воде, вследствие этого нарушается
- 9. 3.Органический йод образуется, но нарушен биосинтез тиреоидных гормонов в ткани железы. Этому ферментативному дефекту соответствует та
- 10. Следовательно, в результате всех причин, три из которых обусловлены мутаций разных генов, развивается одно и то
- 11. Выяснение генетической гетерогенности имеет большое практическое значение: таким образом раскрывается истинный патогенез болезни, ставится правильный диагноз
- 12. Клинический полиморфизм заключается в различиях клинической картины при одном и том же заболеваний. Проявляется он при
- 13. Классификация моногенных болезней В основе клинической классификации лежит органный и системный принципы. Выделяют: 1)Наследственные болезни 2)Психические
- 14. Если систематизировать моногенные болезни по этиологическому принципу, можно выделить болезни: 1)С выясненным биохимическим дефектом 2)С не
- 15. Эти заболевания можно классифицировать по типу передачи патологического признака: 1)Аутосомно-доминантные 2)Аутосомно-рецессивные
- 16. В зависимости от преимущественного поражения вида обмена можно выделить следующие большие группы заболеваний: 1)Наследственные дефекты обмена
- 17. Болезни обмена углеводов 1.Болезнь Помпе 2.Болезнь Гирке 3. Болезнь Мак-Ардля Эти болезни часто называют синдромными
- 18. Болезнь Помпе Заболевание связано с недостаточностью фермента а-глюкозидазы , который отвечает за разложение избытка гликогена, сложной
- 19. Болезнь Помпе
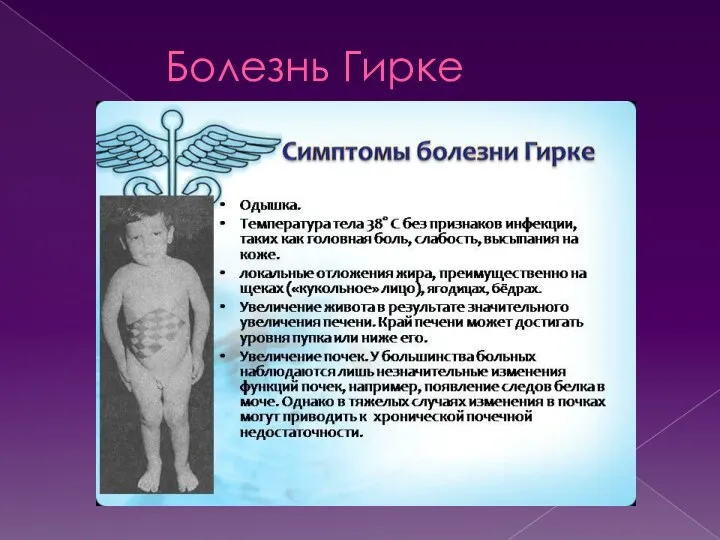
- 20. Болезнь Гирке Болезнь Гирке — гликогеноз (гликогеновая болезнь) вызванная недостаточностью глюкозо-6-фосфатазы. При болезни Гирке сохраняется способность
- 21. Болезнь Гирке
- 22. Болезнь Мак-Ардля Болезнь Мак-Ардля — гликогеноз, связанный с дефектом мышечной фосфорилазы. Заболевание, обусловленное нарушением каталитической функции
- 23. Болезнь Мак-Ардля
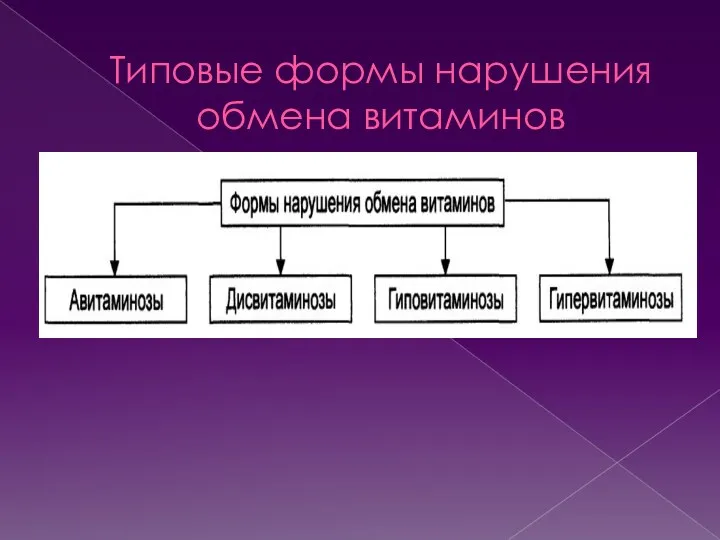
- 24. Типовые формы нарушения обмена витаминов
- 25. Авитаминоз Авитаминоз- патологическое состояние, развивающееся вследствие отсутствия в организме витамина и /или невозможности реализации его эффектов.
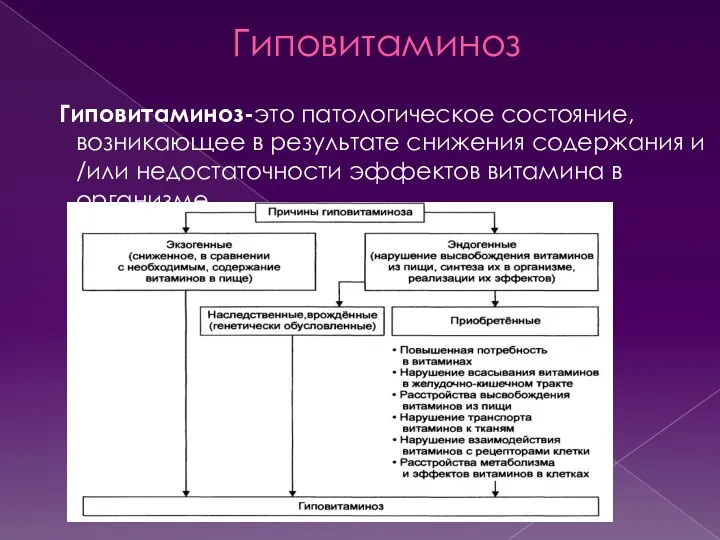
- 26. Гиповитаминоз Гиповитаминоз-это патологическое состояние, возникающее в результате снижения содержания и /или недостаточности эффектов витамина в организме.
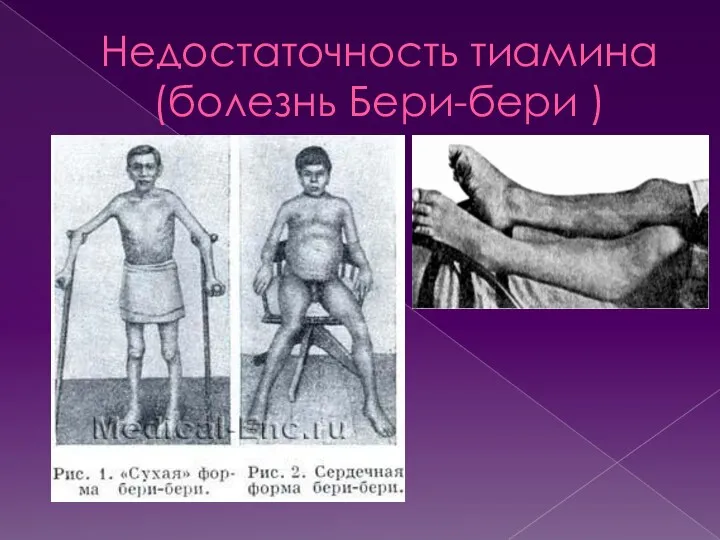
- 27. Недостаточность тиамина (болезнь Бери-бери ) Взрослому человеку необходимо не менее 1,4-2,4 мг витамина B1 в день.
- 28. Недостаточность тиамина (болезнь Бери-бери )
- 29. НЕДОСТАТОЧНОСТЬ ВИТАМИНА D (РАХИТ) Суточная потребность в витамине у детей выше, чем у взрослых, и составляет
- 30. РАХИТ
- 31. Цинга Минимальная потребность взрослого человека в витамине С оценивается в 50-100 мг/сутки. Цинга (скорбут, детская форма
- 32. Цинга
- 33. ПЕЛЛАГРА Пеллагра —заболевание, один из гиповитаминозов, который является следствием длительного неполноценного питания(недостаток витамина РР )и белков.
- 34. Можно отметить ряд особенностей наследственных болезней обмена веществ. Они проявляются рано, часто в детском возрасте. Клиническая
- 35. Патогенез сводится с одной стороны к дефициту образования тех или иных биологических активных веществ или к
- 36. Общим признаком наследственных болезней обмена аминокислот являются: кожные нарушения, снижение интеллекта, судороги, разнообразная неврологическая симптоматика, порезы,
- 37. Группа наследственно-обусловленных мукополисахаридов характеризуется поражением опорно-двигательного аппарата, глаз, сердечено-сосудистой системы и снижением интеллекта.Отмечаются контрактура суставов,уплощенная переносица,карликовость.
- 38. Наследственные болезни обмена минералов включают 2 основные формы: Пароксизмальный паралич(В основе этой наследственной болезни лежит нарушение
- 39. Гепатоцеребральная дистрофия Синонимы-гепатолентикулярная дегенерация, болезнь Вестфаля-Вильсона-Коновалова. Гцд- хроническое прогрессирующее наследственно-дегенеративное заболевание, характеризующееся сочетанным поражением подкорковых узлов
- 40. Гепатоцеребральная дистрофия
- 41. Этиология и патогенез Наследственное заболевание с аутосомно-рецессивным типом наследования. Заболевание генетически обусловлено нарушением синтеза белка церулоплазмина,
- 42. Кольцо Кайзера-Флейшера обнаруживаются при хроническом отравлении медью (например, при кормлении детей молоком, нагретым в медной посуде
- 43. Пароксизмальный паралич Группа нервно-мышечных заболеваний, характеризующихся внезапными приступами мышечной слабости и плегиями. Патогенез не ясен.
- 44. Клинические формы параксизмального паралича: 1.Гипокалиемическая. 2.Гиперкалиемическая. 3.Нормокалиемическая.
- 45. 1.Гипокалиемическая(болезнь Вестфаля). Наследуется как по аутосомно-доминантному, так и по аутосомно –рецессивному типу. Проявляется в 6-15 лет.
- 46. Гипокалиемическая(болезнь Вестфаля)
- 47. 2.Гиперкалиемическая(болезнь Гамсторп). Наследуется по аутосомно-доминантному типу. Проявляется в возрасте 1-5 лет. В отличии от гипокалиемического развивается
- 48. Гиперкалиемическая(болезнь Гамсторп)
- 49. 3.Нормокалиемический(периодический) паралич. Наследуется по аутосомно-доминантному типу. Провляется до 10-летнего возраста. Особенностью её является сравнительно медленно пароксизмально
- 50. Периодический паралич
- 51. Все формы пароксизмальных миоплегий имеют медленно прогрессирующее течение. Прогноз при своевременно поставленном диагнозе, проведении экстренных мероприятий
- 52. Большая группа заболеваний – это наследственные болезни с дефектами эндокринной системы. К ним относятся: 1)Адреро-генитальный синдром-Аномалии
- 53. Селективный скрининг 1.Задержка психомоторного развития у детей раннего возраста( умственная отсталость у детей старшего возраста) 2.Неврологические
- 55. Скачать презентацию

Основные свойства моногенных болезней
1)Менделирующий характер наследования
2)Хронический крогра
3)Генетическая гетерогенность
4) Клинический полиморфизм
Основным
Основные свойства моногенных болезней
1)Менделирующий характер наследования
2)Хронический крогра
3)Генетическая гетерогенность
4) Клинический полиморфизм
Основным

Моногенные болезни можно разделить по типу наследования
1)Аутосомно-доминантные
2)Аутосомно-рецессивные
3)Х -
Моногенные болезни можно разделить по типу наследования
1)Аутосомно-доминантные
2)Аутосомно-рецессивные
3)Х -

Никакие другие группы наследственной патологии (хромосомные и мультифакториальные болезни) таким
Никакие другие группы наследственной патологии (хромосомные и мультифакториальные болезни) таким

Моногенные болезни
это заболевания, протекающие с постоянным прогрессированием патологического процесса.
Моногенные болезни
это заболевания, протекающие с постоянным прогрессированием патологического процесса.

Генетическая гетерогенность заключается в том, что развитие сходного фенотипа (то
Генетическая гетерогенность заключается в том, что развитие сходного фенотипа (то

Генетическую гетерогенность можно рассмотреть на примере врожденного гипотиреоза.Недостаточность продукции
Генетическую гетерогенность можно рассмотреть на примере врожденного гипотиреоза.Недостаточность продукции

1.Поступление неорганического йода в организм снижено из-за недостатка его в
1.Поступление неорганического йода в организм снижено из-за недостатка его в

3.Органический йод образуется, но нарушен биосинтез тиреоидных гормонов в ткани железы.

Следовательно, в результате всех причин, три из которых обусловлены мутаций
Следовательно, в результате всех причин, три из которых обусловлены мутаций

Выяснение генетической гетерогенности имеет большое практическое значение: таким образом раскрывается
Выяснение генетической гетерогенности имеет большое практическое значение: таким образом раскрывается

Клинический полиморфизм заключается в различиях клинической картины при одном и
Клинический полиморфизм заключается в различиях клинической картины при одном и

Классификация моногенных болезней
В основе клинической классификации лежит органный и
Классификация моногенных болезней В основе клинической классификации лежит органный и

Если систематизировать моногенные болезни по этиологическому принципу, можно выделить болезни:
1)С выясненным
1)С выясненным

Эти заболевания можно
классифицировать по типу передачи
патологического признака:
1)Аутосомно-доминантные
2)Аутосомно-рецессивные
классифицировать по типу передачи
патологического признака:
1)Аутосомно-доминантные
2)Аутосомно-рецессивные

В зависимости от преимущественного поражения вида обмена можно выделить следующие большие
В зависимости от преимущественного поражения вида обмена можно выделить следующие большие

Болезни обмена углеводов
1.Болезнь Помпе
2.Болезнь Гирке
3. Болезнь Мак-Ардля
Эти болезни часто называют
Болезни обмена углеводов
1.Болезнь Помпе
2.Болезнь Гирке
3. Болезнь Мак-Ардля
Эти болезни часто называют

Болезнь Помпе
Заболевание связано с недостаточностью фермента а-глюкозидазы , который
Болезнь Помпе
Заболевание связано с недостаточностью фермента а-глюкозидазы , который

Болезнь Помпе
Болезнь Помпе

Болезнь Гирке
Болезнь Гирке — гликогеноз (гликогеновая болезнь) вызванная недостаточностью глюкозо-6-фосфатазы. При болезни
Болезнь Гирке
Болезнь Гирке — гликогеноз (гликогеновая болезнь) вызванная недостаточностью глюкозо-6-фосфатазы. При болезни
Болезнь Гирке
Болезнь Гирке

Болезнь Мак-Ардля
Болезнь Мак-Ардля — гликогеноз, связанный с дефектом мышечной фосфорилазы.
Болезнь Мак-Ардля
Болезнь Мак-Ардля — гликогеноз, связанный с дефектом мышечной фосфорилазы.

Болезнь Мак-Ардля
Болезнь Мак-Ардля
Типовые формы нарушения обмена витаминов
Типовые формы нарушения обмена витаминов

Авитаминоз
Авитаминоз- патологическое состояние, развивающееся вследствие отсутствия в организме витамина и /или
Авитаминоз
Авитаминоз- патологическое состояние, развивающееся вследствие отсутствия в организме витамина и /или
Гиповитаминоз
Гиповитаминоз-это патологическое состояние, возникающее в результате снижения содержания и /или
Гиповитаминоз
Гиповитаминоз-это патологическое состояние, возникающее в результате снижения содержания и /или

Недостаточность тиамина (болезнь Бери-бери )
Взрослому человеку необходимо не менее 1,4-2,4 мг
Недостаточность тиамина (болезнь Бери-бери )
Взрослому человеку необходимо не менее 1,4-2,4 мг
Недостаточность тиамина (болезнь Бери-бери )
Недостаточность тиамина (болезнь Бери-бери )

НЕДОСТАТОЧНОСТЬ ВИТАМИНА D (РАХИТ)
Суточная потребность в витамине у детей выше, чем
НЕДОСТАТОЧНОСТЬ ВИТАМИНА D (РАХИТ)
Суточная потребность в витамине у детей выше, чем
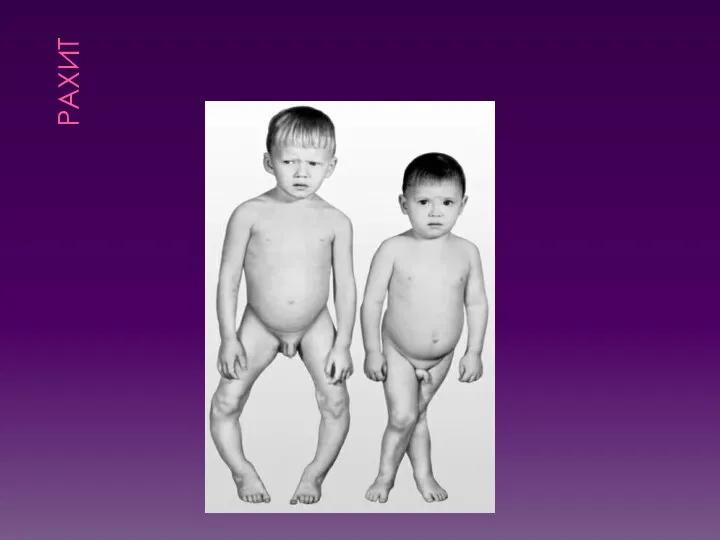
РАХИТ
РАХИТ
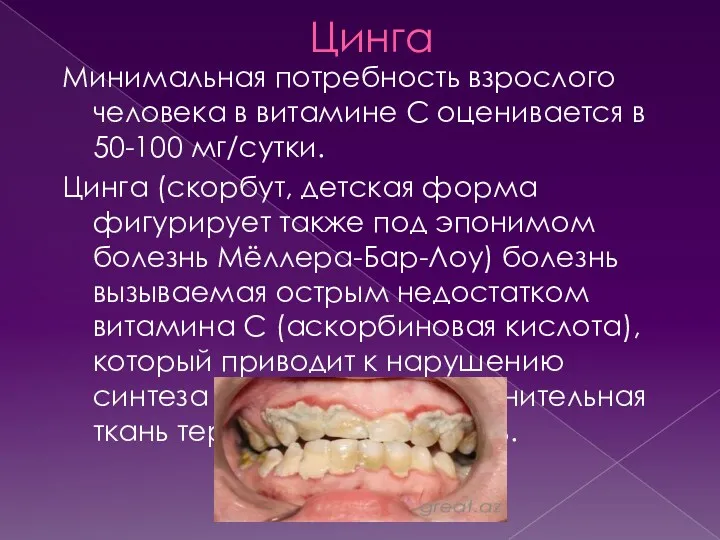
Цинга
Минимальная потребность взрослого человека в витамине С оценивается в 50-100 мг/сутки.
Цинга
Цинга
Минимальная потребность взрослого человека в витамине С оценивается в 50-100 мг/сутки.
Цинга
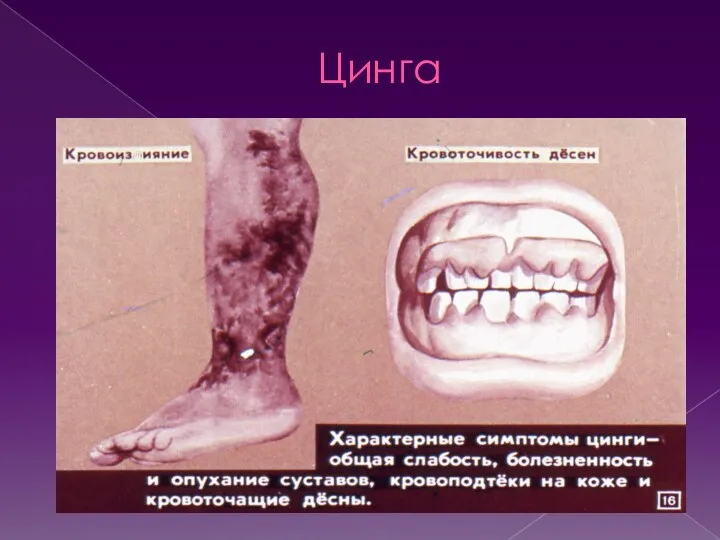
Цинга
Цинга

ПЕЛЛАГРА
Пеллагра —заболевание, один из гиповитаминозов, который является следствием длительного неполноценного питания(недостаток витамина
ПЕЛЛАГРА
Пеллагра —заболевание, один из гиповитаминозов, который является следствием длительного неполноценного питания(недостаток витамина

Можно отметить ряд особенностей наследственных болезней обмена веществ. Они проявляются
Можно отметить ряд особенностей наследственных болезней обмена веществ. Они проявляются

Патогенез сводится с одной стороны к дефициту образования тех или
Патогенез сводится с одной стороны к дефициту образования тех или

Общим признаком наследственных болезней обмена аминокислот являются: кожные нарушения, снижение интеллекта,

Группа наследственно-обусловленных мукополисахаридов характеризуется поражением опорно-двигательного аппарата, глаз, сердечено-сосудистой системы
Группа наследственно-обусловленных мукополисахаридов характеризуется поражением опорно-двигательного аппарата, глаз, сердечено-сосудистой системы

Наследственные болезни обмена минералов включают 2 основные формы: Пароксизмальный паралич(В основе
Наследственные болезни обмена минералов включают 2 основные формы: Пароксизмальный паралич(В основе

Гепатоцеребральная дистрофия
Синонимы-гепатолентикулярная дегенерация, болезнь Вестфаля-Вильсона-Коновалова.
Гцд- хроническое прогрессирующее наследственно-дегенеративное заболевание, характеризующееся сочетанным
Гепатоцеребральная дистрофия
Синонимы-гепатолентикулярная дегенерация, болезнь Вестфаля-Вильсона-Коновалова.
Гцд- хроническое прогрессирующее наследственно-дегенеративное заболевание, характеризующееся сочетанным
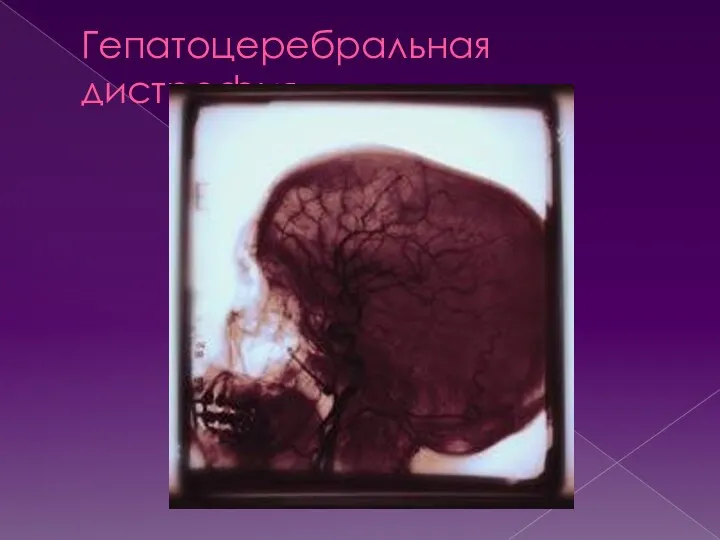
Гепатоцеребральная дистрофия
Гепатоцеребральная дистрофия
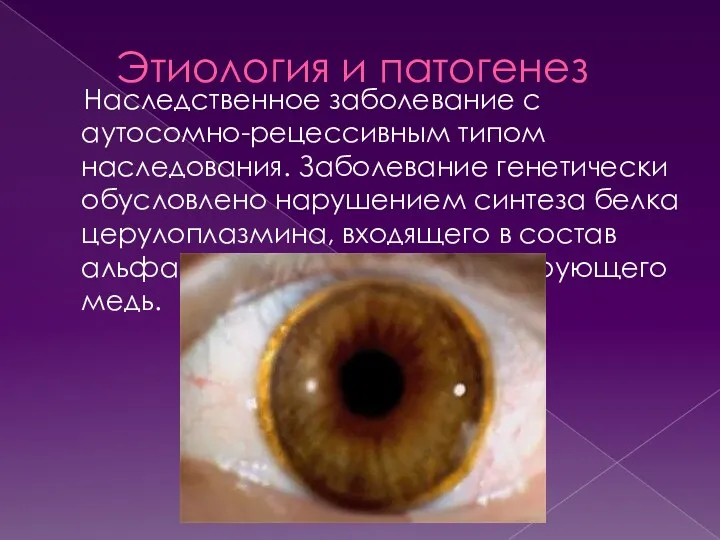
Этиология и патогенез
Наследственное заболевание с аутосомно-рецессивным типом наследования. Заболевание
Этиология и патогенез
Наследственное заболевание с аутосомно-рецессивным типом наследования. Заболевание
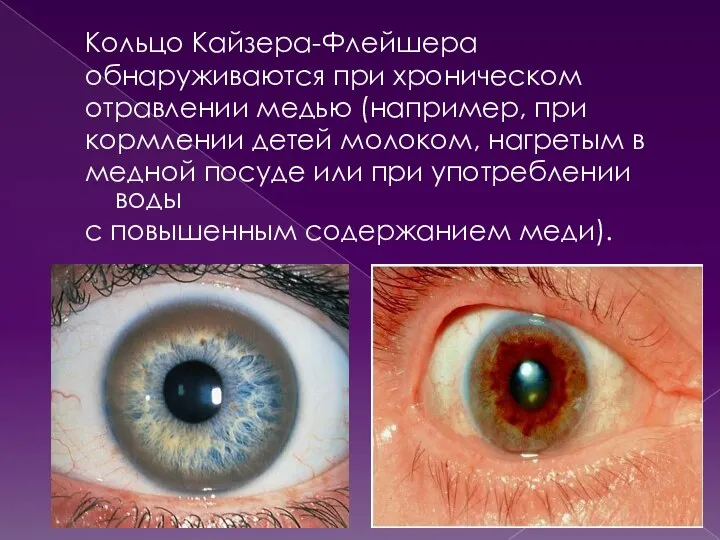
Кольцо Кайзера-Флейшера
обнаруживаются при хроническом
отравлении медью (например, при
кормлении детей молоком, нагретым в
медной
Кольцо Кайзера-Флейшера
обнаруживаются при хроническом
отравлении медью (например, при
кормлении детей молоком, нагретым в
медной

Пароксизмальный паралич
Группа нервно-мышечных заболеваний, характеризующихся внезапными приступами мышечной слабости и
Пароксизмальный паралич
Группа нервно-мышечных заболеваний, характеризующихся внезапными приступами мышечной слабости и

Клинические формы параксизмального паралича:
1.Гипокалиемическая.
2.Гиперкалиемическая.
3.Нормокалиемическая.
Клинические формы параксизмального паралича:
1.Гипокалиемическая.
2.Гиперкалиемическая.
3.Нормокалиемическая.

1.Гипокалиемическая(болезнь Вестфаля).
Наследуется как по аутосомно-доминантному, так и по аутосомно
1.Гипокалиемическая(болезнь Вестфаля).
Наследуется как по аутосомно-доминантному, так и по аутосомно

Гипокалиемическая(болезнь Вестфаля)
Гипокалиемическая(болезнь Вестфаля)

2.Гиперкалиемическая(болезнь Гамсторп).
Наследуется по аутосомно-доминантному типу. Проявляется в возрасте 1-5 лет.
2.Гиперкалиемическая(болезнь Гамсторп).
Наследуется по аутосомно-доминантному типу. Проявляется в возрасте 1-5 лет.
Гиперкалиемическая(болезнь Гамсторп)
Гиперкалиемическая(болезнь Гамсторп)

3.Нормокалиемический(периодический) паралич.
Наследуется по аутосомно-доминантному типу. Провляется до 10-летнего возраста.
3.Нормокалиемический(периодический) паралич.
Наследуется по аутосомно-доминантному типу. Провляется до 10-летнего возраста.
Периодический паралич
Периодический паралич

Все формы пароксизмальных миоплегий имеют медленно прогрессирующее течение.
Прогноз при своевременно
Все формы пароксизмальных миоплегий имеют медленно прогрессирующее течение.
Прогноз при своевременно

Большая группа заболеваний – это наследственные болезни с дефектами эндокринной системы.
Большая группа заболеваний – это наследственные болезни с дефектами эндокринной системы.

Селективный скрининг
1.Задержка психомоторного развития у детей раннего возраста( умственная отсталость
Селективный скрининг
1.Задержка психомоторного развития у детей раннего возраста( умственная отсталость
 Тамақтан улану кезіндегі алғашқы жәрдем
Тамақтан улану кезіндегі алғашқы жәрдем Бауырдың патологиялық процестері
Бауырдың патологиялық процестері Медико-демографические показатели населения Горноуральского городского округа
Медико-демографические показатели населения Горноуральского городского округа Заболевания легких у беременных
Заболевания легких у беременных Расспрос пациента
Расспрос пациента Фізіологія системи крові. Еритрон. Групи крові
Фізіологія системи крові. Еритрон. Групи крові Современные генетические технологии в медицине
Современные генетические технологии в медицине Врожденные черепно - мозговые грыжи
Врожденные черепно - мозговые грыжи Лимфогранулематоз бен лимфолейкоз кезіндегі лимфа түйіндердің ұлғаю ерекшеліктері
Лимфогранулематоз бен лимфолейкоз кезіндегі лимфа түйіндердің ұлғаю ерекшеліктері Профессиональные вредности и профессиональные заболевания и их профилактика в инфекционном отделении
Профессиональные вредности и профессиональные заболевания и их профилактика в инфекционном отделении Патогенные представители семейства энтеробактерий - возбудители шигеллезов, сальмонеллезов. Патогенные кишечные палочки
Патогенные представители семейства энтеробактерий - возбудители шигеллезов, сальмонеллезов. Патогенные кишечные палочки Шизофрения. Физиология заболевания. Гипотеза Фанберга
Шизофрения. Физиология заболевания. Гипотеза Фанберга Жас балалардағы тырнақ және шаш ерекшіліктері
Жас балалардағы тырнақ және шаш ерекшіліктері Дәрілік өсімдіктерді ресурстанулық зерттеу. Қазақстан өсімдіктері және дәрілік өсімдіктердің таралуы
Дәрілік өсімдіктерді ресурстанулық зерттеу. Қазақстан өсімдіктері және дәрілік өсімдіктердің таралуы Состояние условий труда на предприятиях Пермского края. Сохранение здоровья работающих
Состояние условий труда на предприятиях Пермского края. Сохранение здоровья работающих Отравления. Укусы животных и насекомых
Отравления. Укусы животных и насекомых Primary and secondary tuberculosis. (Lecture 5)
Primary and secondary tuberculosis. (Lecture 5) Сердечно-сосудистая хирургия
Сердечно-сосудистая хирургия Шизофрения. Особенности в детском возрасте
Шизофрения. Особенности в детском возрасте Вирусные гепатиты: лечение и профилактика
Вирусные гепатиты: лечение и профилактика Дифтерия. Возбудители - бактерии Леффлера
Дифтерия. Возбудители - бактерии Леффлера Поражения слизистой оболочки полости рта у детей, обусловленные аллергией
Поражения слизистой оболочки полости рта у детей, обусловленные аллергией Болезни век и слезных органов
Болезни век и слезных органов Гнойно-воспалительные заболевания кожи и подкожной клетчатки у детей
Гнойно-воспалительные заболевания кожи и подкожной клетчатки у детей Механизмы репарации ДНК
Механизмы репарации ДНК Предмет, задачи и методы патологии
Предмет, задачи и методы патологии Лекарственные средства, стимулирующие цнс: психостимуляторы, ноотропы аналептики, антидепрессанты
Лекарственные средства, стимулирующие цнс: психостимуляторы, ноотропы аналептики, антидепрессанты Туберкулез внелегочной локализации
Туберкулез внелегочной локализации