- Мукополисахаридоз. Биологическое значение

Содержание
- 2. Мукополисахаридо́зы группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов (GAG, мукополисахаридов), связанных недостаточностью
- 3. Биологическое значение Гликозаминогликанов Гликозаминогликаны в составе протеогликанов входят в состав межклеточного вещества соединительной ткани, содержатся в
- 4. Лизосо́мные боле́зни накопле́ния общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом.
- 5. Классификация укмополисахаридозов Современная классификация, в зависимости от характера ферментативного дефекта, выделяет несколько основных типов укмополисахаридозов: I
- 6. Синдром Гурлера Клиническая картина Дети с синдромом Гурлер отличаются низкорослостью (отставание в физическом развитии наблюдается с
- 8. Синдром Шейе Клиническая картина Синдром Шейе (мукополисахаридоз-I S) является менее тяжёлым вариантом синдрома Гурлер. Клиническая симптоматика
- 9. Синдром Гурлер — Шейе Клиническая картина Синдром Гурлер — Шейе (мукополисахаридоз-I H/S) является менее тяжёлым промежуточным
- 10. Болезнь Хантера Болезнь проявляется в раннем возрасте (2—4 года) утолщением ноздрей, губ, языка, тугоподвижностью суставов, задержкой
- 12. Симптомы Мукополисахаридоза типа III (синдрома Санфилиппо, болезни Санфилиппо) Заболевание манифестирует обычно на 2-м году жизни ребенка;
- 14. Синдром Моркио Проявляется с 2-летнего возраста. Болезнь характеризуется карликовостью (рост взрослого больного около 100 см), непропорциональным
- 16. Синдром Марото — Лами Интеллектуальное развитие детей с синдром Марото — Лами, как правило, не страдает
- 17. Синдром Слая Характерными проявлениями синдрома являются: паховые и пупочные грыжи, низкий рост, килевидная грудная клетка, кифоз,
- 18. Патогенез В зависимости от недостаточности одного из ферментов лизосом, накапливаются мукополисахариды одного из трёх классов: гепаран-,
- 19. Как любое другое Х-сцепленное рецессивное заболевание, Болезнь Хантера наследуется следующим образом.
- 21. Скачать презентацию

Мукополисахаридо́зы
группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов (GAG, мукополисахаридов), связанных недостаточностью
Мукополисахаридо́зы
группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов (GAG, мукополисахаридов), связанных недостаточностью

Биологическое значение Гликозаминогликанов
Гликозаминогликаны в составе протеогликанов входят в состав межклеточного вещества
Биологическое значение Гликозаминогликанов
Гликозаминогликаны в составе протеогликанов входят в состав межклеточного вещества

Лизосо́мные боле́зни накопле́ния
общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции
Лизосо́мные боле́зни накопле́ния
общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции

Классификация укмополисахаридозов
Современная классификация, в зависимости от характера ферментативного дефекта, выделяет несколько основных типов
Классификация укмополисахаридозов
Современная классификация, в зависимости от характера ферментативного дефекта, выделяет несколько основных типов

Синдром Гурлера
Клиническая картина
Дети с синдромом Гурлер отличаются низкорослостью (отставание в физическом развитии наблюдается
Синдром Гурлера
Клиническая картина
Дети с синдромом Гурлер отличаются низкорослостью (отставание в физическом развитии наблюдается


Синдром Шейе
Клиническая картина
Синдром Шейе (мукополисахаридоз-I S) является менее тяжёлым вариантом синдрома Гурлер.
Синдром Шейе
Клиническая картина
Синдром Шейе (мукополисахаридоз-I S) является менее тяжёлым вариантом синдрома Гурлер.

Синдром Гурлер — Шейе
Клиническая картина
Синдром Гурлер — Шейе (мукополисахаридоз-I H/S) является менее
Синдром Гурлер — Шейе
Клиническая картина
Синдром Гурлер — Шейе (мукополисахаридоз-I H/S) является менее

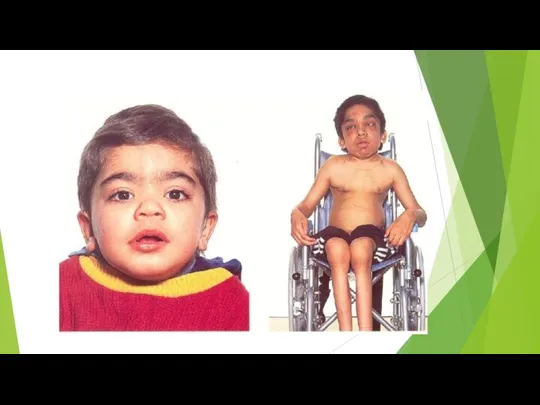
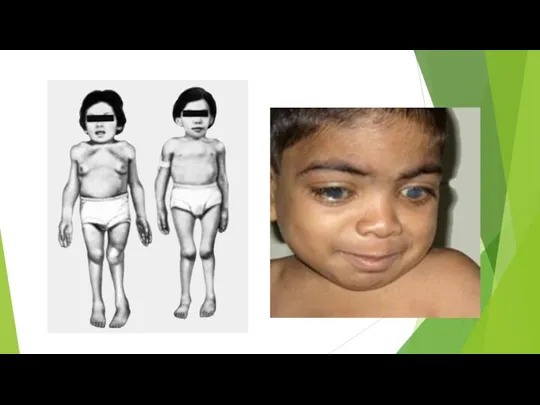
Болезнь Хантера
Болезнь проявляется в раннем возрасте (2—4 года) утолщением ноздрей, губ,
Болезнь Хантера
Болезнь проявляется в раннем возрасте (2—4 года) утолщением ноздрей, губ,


Симптомы Мукополисахаридоза типа III (синдрома Санфилиппо, болезни Санфилиппо)
Заболевание манифестирует обычно на
Симптомы Мукополисахаридоза типа III (синдрома Санфилиппо, болезни Санфилиппо)
Заболевание манифестирует обычно на

Синдром Моркио
Проявляется с 2-летнего возраста. Болезнь характеризуется карликовостью (рост взрослого больного
Синдром Моркио
Проявляется с 2-летнего возраста. Болезнь характеризуется карликовостью (рост взрослого больного

Синдром Марото — Лами
Интеллектуальное развитие детей с синдром Марото — Лами, как
Синдром Марото — Лами
Интеллектуальное развитие детей с синдром Марото — Лами, как

Синдром Слая
Характерными проявлениями синдрома являются: паховые и пупочные грыжи, низкий рост,
Синдром Слая
Характерными проявлениями синдрома являются: паховые и пупочные грыжи, низкий рост,

Патогенез
В зависимости от недостаточности одного из ферментов лизосом, накапливаются мукополисахариды одного из трёх классов: гепаран-, дерматан- или кератансульфаты
Патогенез
В зависимости от недостаточности одного из ферментов лизосом, накапливаются мукополисахариды одного из трёх классов: гепаран-, дерматан- или кератансульфаты
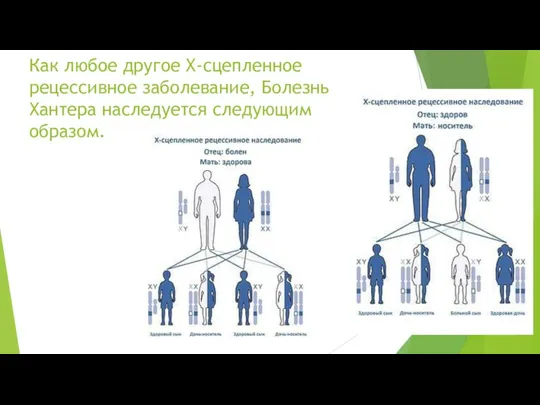
Как любое другое Х-сцепленное рецессивное заболевание, Болезнь Хантера наследуется следующим образом.
Как любое другое Х-сцепленное рецессивное заболевание, Болезнь Хантера наследуется следующим образом.
 Загар-польза или вред
Загар-польза или вред Физиология сердца. Физиологические основы гемодинамики
Физиология сердца. Физиологические основы гемодинамики Магнитно-резонансная томография (МРТ) и функциональная магнитно-резонансная томография (фМРТ)
Магнитно-резонансная томография (МРТ) и функциональная магнитно-резонансная томография (фМРТ) Первая помощь при кровотечении
Первая помощь при кровотечении Аллергический ринит. Острый экзогенный аллергический альвеолит
Аллергический ринит. Острый экзогенный аллергический альвеолит Процесс терморегуляции
Процесс терморегуляции Протездерді өңдеу технологиясы. Абразивті материалдар
Протездерді өңдеу технологиясы. Абразивті материалдар Йод. Естественные источники йода в рационе человека
Йод. Естественные источники йода в рационе человека Ішкі сәулеленуден медициналық қорғану
Ішкі сәулеленуден медициналық қорғану Характеристики электроэнцефалограммы при наиболее распространённых формах эпилепсии и эпилептических синдромов
Характеристики электроэнцефалограммы при наиболее распространённых формах эпилепсии и эпилептических синдромов History of medicine as science and subject for study. Prehistoric medicine
History of medicine as science and subject for study. Prehistoric medicine Методы осмотра и исследования ЛОР-органов
Методы осмотра и исследования ЛОР-органов Иммунотропные средства
Иммунотропные средства Атеросклероз – одно из самых актуальных заболеваний ХХ века
Атеросклероз – одно из самых актуальных заболеваний ХХ века Гигиена сердечно - сосудистой системы (8 класс)
Гигиена сердечно - сосудистой системы (8 класс) Тыныс алу жолдарының өткізгіштігін қалыпқа келтіру әдістері, көрсеткіштері мен асқынулары
Тыныс алу жолдарының өткізгіштігін қалыпқа келтіру әдістері, көрсеткіштері мен асқынулары Индивидуальная гигиена полости рта
Индивидуальная гигиена полости рта Сестринская помощь при остром инфаркте миокарда
Сестринская помощь при остром инфаркте миокарда Явления шока
Явления шока Реабилитация. Медицинская реабилитация
Реабилитация. Медицинская реабилитация Нашақорлық пен токсикомания эпидемиологиясы. Тәуелділік аурулары кезіндегі соматикалық бұзылыстар
Нашақорлық пен токсикомания эпидемиологиясы. Тәуелділік аурулары кезіндегі соматикалық бұзылыстар Предмет и задачи судебной психиатрии
Предмет и задачи судебной психиатрии Значение открытий в области физики для развития медицины
Значение открытий в области физики для развития медицины Ведение беременных с одной почкой
Ведение беременных с одной почкой Сальмонеллез
Сальмонеллез Определение комы
Определение комы Медицина катастроф
Медицина катастроф Синдром приобретённого иммунного дефицита (СПИД)
Синдром приобретённого иммунного дефицита (СПИД)