- Наследственно-дегенеративные заболевания нервной системы

Содержание
- 2. Наследственно-дегенеративные заболевания нервной системы – обширная группа болезней, обусловленных изменениями генетической информации (генные мутации). Генные мутации
- 3. Классификация: I. Миопатии: 1. Мышечная дистрофия Дюшенна. 2. Миопатия Эрба (конечностно-поясная форма). 3. Лице-лопаточно-плечевая форма Ландузи-Дежерина.
- 4. II.а Спинальные амиотрофии: 1. Спинальная амиотрофия Вердинга-Гоффманна. 2. Прогрессирующая спинальная амиотрофия Кугельберга-Веландера. II.б Невральные амиотрофии. Невральная
- 5. III. Миастения. IV. Миатония. Врожденная миотония (Болезнь Оппенгейма). V. Миотония Томсона. VI. Пароксизмальная миоплегия.
- 6. Патоморфология миопатий: Нарушение распределения типов мышечных волокон. Изменение размера мышечных волокон. Нарушение строения мышечных волокон. Патологические
- 7. Патоморфология амиотрофий: Грубое поражение клеток передних рогов, передних корешков и периферических нервов спинного мозга: уменьшение количества
- 8. Патоморфология миастении: 1. Нарушение синаптического проведения импульсов. 2. Эндокринные расстройства, особенно дисфункция вилочковой железы. 3. В
- 9. Патоморфология миотонии: Недоразвитие клеток передних рогов спинного мозга. Дегенеративные изменения мышечных волокон.
- 10. Патогенез пароксизмальной миоплегии: Нарушение синаптической передачи и недостаточная генерация потенциала концевых пластинок. Нарушение распространения потенциала действия
- 11. Мышечная дистрофия Дюшенна: Начало заболевания в раннем возрасте – 3 – 4 года. Преимущественно поражение мышц
- 12. Сухожильные рефлексы вначале снижаются, затем исчезают. Отмечаются атрофия мышц языка, мягкого нёба, гортани и жевательных мышц.
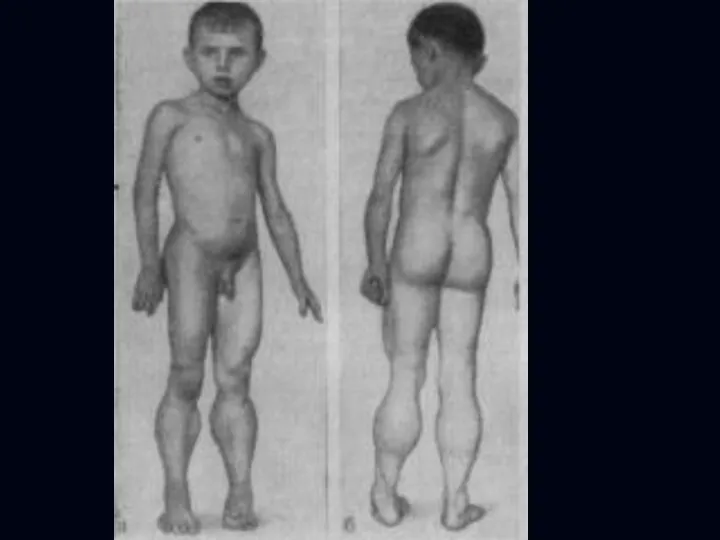
- 15. Юношеская миопатия Эрба-Рота: Наследуется по аутосомно-рецессивному типу (болеют дети здоровых родителей). Болезнь начинается в возрасте от
- 16. Больные испытывают затруднение при вставании с пола, помогая себе при этом руками; по ступенькам лестницы поднимаются,
- 25. Лице-лопаточно-плечевая форма Ландузи-Дежерина. Начало заболевания с 7 - 15 лет. Слабость и атрофии мышц лица и
- 26. Симптом «крыловидных лопаток», горизонтальное расположение ключиц. Характерны толстые, выпяченные вперед губы («губы тапира»), «поперечная улыбка». Выраженный
- 28. Спинальная амиотрофия Вердига – Гоффмана. Выделяют формы: Врожденная форма. Ранняя детская форма. Поздняя форма.
- 29. Врожденная форма: Начало внутриутробно. Слабое шевеление плода. При рождении вялый паралич в проксимальных отделах конечностей. Бульбарные
- 30. Ранняя детская форма: Начало до 1,5 лет. После интеркурретного заболевания ребенок начинает терять приобретенные навыки. Вялые
- 31. Поздняя форма: Начало в 1,5 – 2 года. Вялые параличи проксимальных отделов ног, затем рук. Мышечные
- 36. Спинальная амиотрофия Кугельберга-Веландера (псевдомиопатическая). Тип наследования аутосомно-рецессивный. Начало в 6 лет. Течение доброкачественное, ещё в 30
- 37. Невральная амиотрофия Шарко-Мари Тип наследования аутосомно-доминантный. Начальные проявления с 13-17 лет. Характерны дистальные парезы, атрофии, сухожильная
- 38. Атрофия мышц голеней и нижней трети бедер. Ноги по виду напоминают «ноги аиста». Часто сочетается с
- 40. Миастения: Появляется утомляемость, распространяется на мышцы: губ, жевательные, глазодвигательные, глотательные. Наблюдается птоз, диплопия, поперечная улыбка (симптом
- 41. Утомляемость появляется в мышцах конечностей. Наблюдается увеличение вилочковой железы. Заболевание имеет тенденцию к прогрессированию. Могут возникать
- 42. Врожденная миатония Оппенгейма: С рождения – мало активных движений. Резкая гипотония или полная атония мышц. Отмечается
- 43. Отсутствуют также гипертрофия и псевдогипертрофия. Лицевые мышцы поражаются редко. Психика сохранена. Тазовых и сенсорных нарушений нет.
- 50. Пароксизмальная миоплегия - генетически детерминированные нервно-мышечные заболевания, обусловленные нарушениями обмена калия и характеризующиеся приступами вялого паралича
- 51. Гипокалиемическая форма: 1. Начало от 10 до 16 лет. 2. Возникновение приступов часто связано с циклом
- 52. 6. При парциальном приступе мышечная слабость и парез или плегия развивается на одной конечности, реже в
- 53. Гиперкалиемическая форма: 1. Начало от 10 до 18 лет. 2. Возникают приступы пароксизмаль-ного гиперкалиемического паралича, как
- 54. 5. В последующем развивается мышечная слабость, гипотония мышц, снижение сухожильных рефлексов. 6. Приступ сопровождается выраженными вегетативными
- 55. Нормокалиемическая форма: 1. Начало в первые 10 лет жизни. 2. Сочетание признаков мышечной слабости и ограничения
- 56. Дополнительные методы обследования: Электромиография (ЭМГ). Биопсия скелетной мышцы. Биохимический анализ крови (БАК). Компьютерная томография скелетных мышц.
- 57. Лечение прогрессирующих мышечных миопатий: Для улучшения трофики: АТФ. Антихолинэстеразные препараты: галантамин. Витаминотерапия: витамины группы В, Е.
- 58. Лечение прогрессирующих мышечных амиотрофий: Для улучшения трофики: АТФ, глютаминовая кислота, метионин, лейцин. Антихолинэстеразные препараты: прозерин, галантамин,
- 59. Лечение невральной амиотрофии Шарко-Мари: Антихолинэстеразные препараты: прозерин, дибазол. Для улучшения трофики: АТФ. ЛФК, массаж, четырехкамерные ванны,
- 60. Лечение миастении: Антихолинэстеразные препараты: прозерин, местинон, оксазил. Препарат, увеличивающий нервно-мышечную передачу – хлористый калий. Рентгенотерапия на
- 61. Лечение пароксизмальной миоплегии: Во время приступа. Высокие дозы хлорида калия внутрь (10-15 г в виде раствора)
- 62. В межприступный период: 1) Диета с высоким содержанием калия и ограничением углеводов и натрия. 2) Спиронолактон,
- 64. Скачать презентацию

Наследственно-дегенеративные заболевания нервной системы
– обширная группа болезней, обусловленных изменениями генетической
Наследственно-дегенеративные заболевания нервной системы – обширная группа болезней, обусловленных изменениями генетической

Классификация:
I. Миопатии:
1. Мышечная дистрофия Дюшенна.
2. Миопатия Эрба (конечностно-поясная форма).
Классификация:
I. Миопатии:
1. Мышечная дистрофия Дюшенна.
2. Миопатия Эрба (конечностно-поясная форма).

II.а Спинальные амиотрофии:
1. Спинальная амиотрофия Вердинга-Гоффманна.
2. Прогрессирующая спинальная
II.а Спинальные амиотрофии:
1. Спинальная амиотрофия Вердинга-Гоффманна.
2. Прогрессирующая спинальная

III. Миастения.
IV. Миатония.
Врожденная миотония
(Болезнь Оппенгейма).
V. Миотония Томсона.
VI. Пароксизмальная миоплегия.
III. Миастения.
IV. Миатония.
Врожденная миотония
(Болезнь Оппенгейма).
V. Миотония Томсона.
VI. Пароксизмальная миоплегия.

Патоморфология миопатий:
Нарушение распределения типов мышечных волокон.
Изменение размера мышечных волокон.
Нарушение строения мышечных
Патоморфология миопатий:
Нарушение распределения типов мышечных волокон.
Изменение размера мышечных волокон.
Нарушение строения мышечных

Патоморфология амиотрофий:
Грубое поражение клеток передних рогов, передних корешков и
Патоморфология амиотрофий:
Грубое поражение клеток передних рогов, передних корешков и

Патоморфология миастении:
1. Нарушение синаптического проведения импульсов.
2. Эндокринные расстройства, особенно дисфункция вилочковой
Патоморфология миастении:
1. Нарушение синаптического проведения импульсов.
2. Эндокринные расстройства, особенно дисфункция вилочковой

Патоморфология миотонии:
Недоразвитие клеток передних рогов спинного мозга.
Дегенеративные изменения мышечных волокон.
Патоморфология миотонии:
Недоразвитие клеток передних рогов спинного мозга.
Дегенеративные изменения мышечных волокон.

Патогенез пароксизмальной миоплегии:
Нарушение синаптической передачи и недостаточная генерация потенциала концевых пластинок.
Нарушение
Патогенез пароксизмальной миоплегии:
Нарушение синаптической передачи и недостаточная генерация потенциала концевых пластинок.
Нарушение

Мышечная дистрофия Дюшенна:
Начало заболевания в раннем возрасте – 3 – 4
Мышечная дистрофия Дюшенна:
Начало заболевания в раннем возрасте – 3 – 4

Сухожильные рефлексы вначале снижаются, затем исчезают.
Отмечаются атрофия мышц языка, мягкого нёба,
Отмечаются атрофия мышц языка, мягкого нёба,
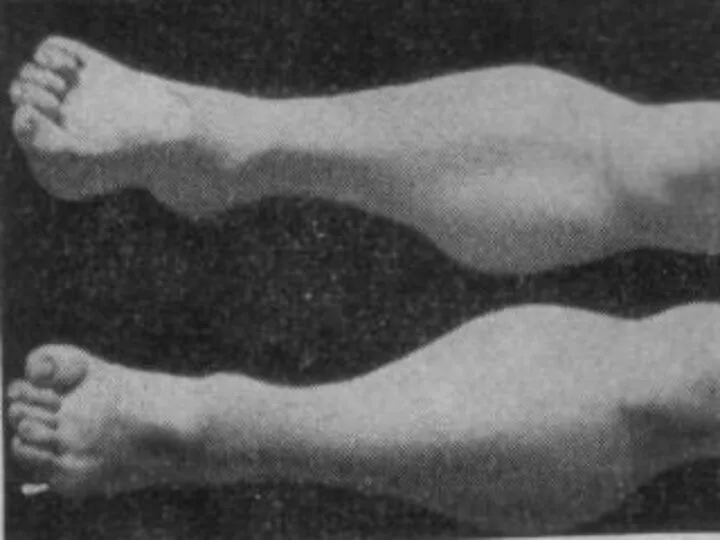

Юношеская миопатия Эрба-Рота:
Наследуется по аутосомно-рецессивному типу (болеют дети здоровых родителей).
Болезнь
Юношеская миопатия Эрба-Рота:
Наследуется по аутосомно-рецессивному типу (болеют дети здоровых родителей).
Болезнь

Больные испытывают затруднение при вставании с пола, помогая себе при этом
Больные испытывают затруднение при вставании с пола, помогая себе при этом
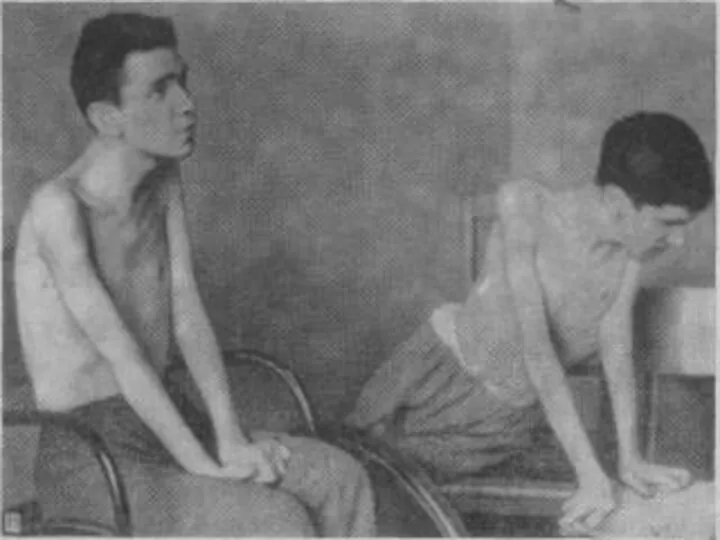
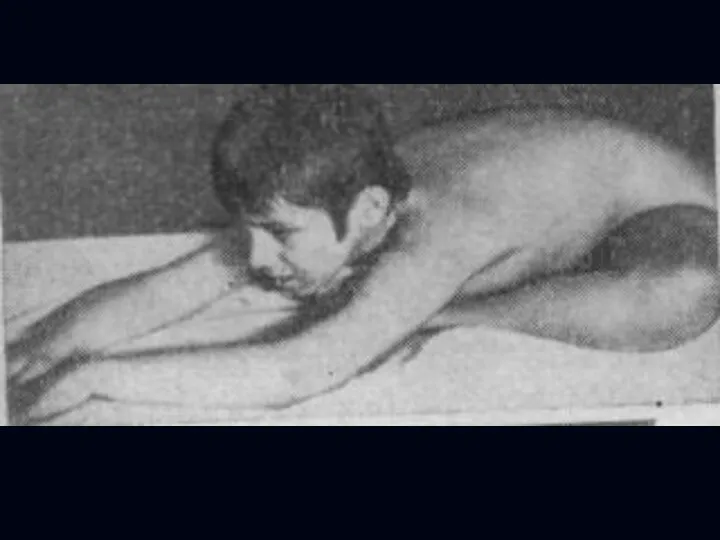
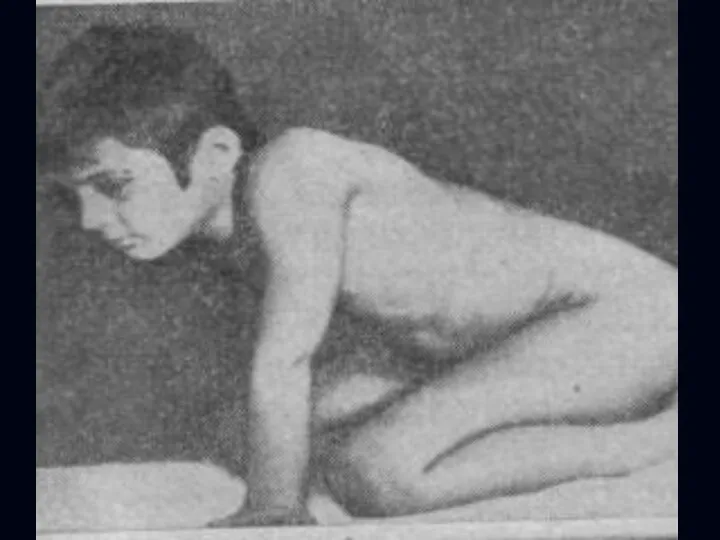

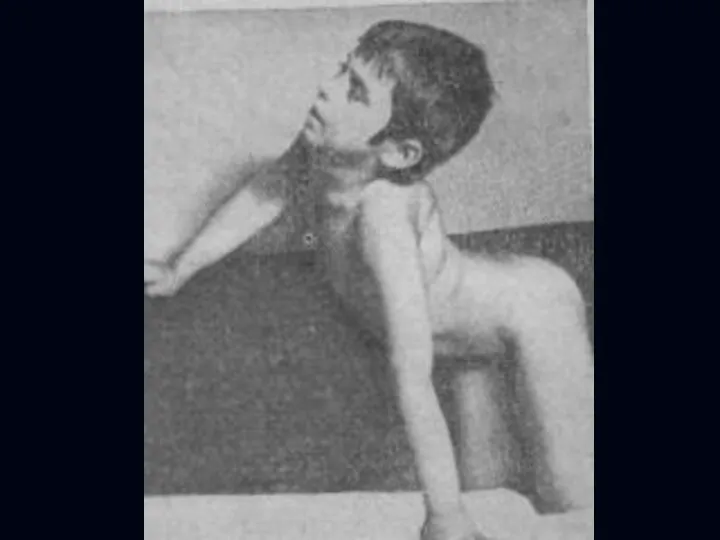

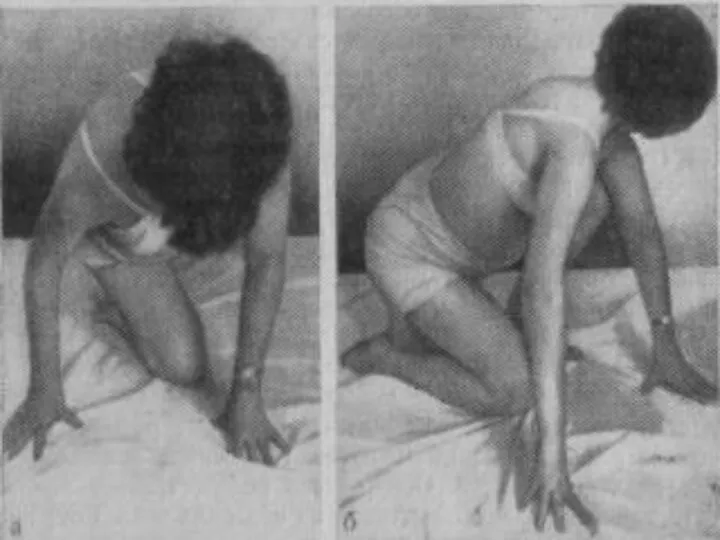
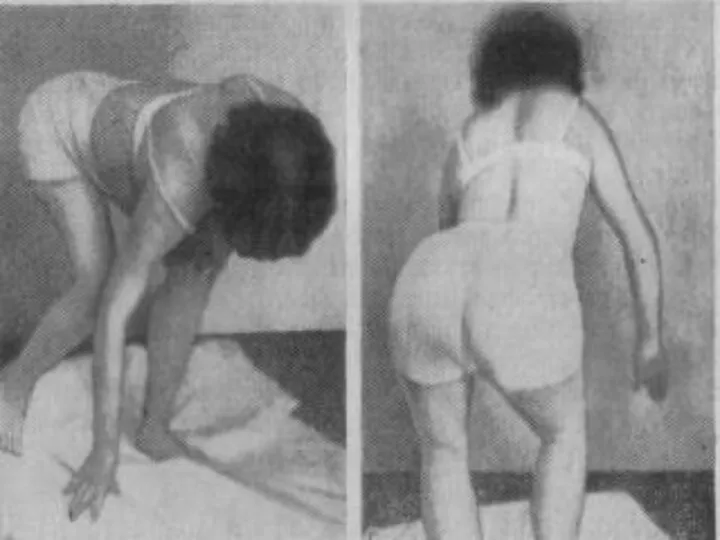

Лице-лопаточно-плечевая форма Ландузи-Дежерина.
Начало заболевания с 7 - 15 лет.
Слабость и атрофии
Лице-лопаточно-плечевая форма Ландузи-Дежерина.
Начало заболевания с 7 - 15 лет.
Слабость и атрофии

Симптом «крыловидных лопаток», горизонтальное расположение ключиц.
Характерны толстые, выпяченные вперед губы («губы
Симптом «крыловидных лопаток», горизонтальное расположение ключиц.
Характерны толстые, выпяченные вперед губы («губы
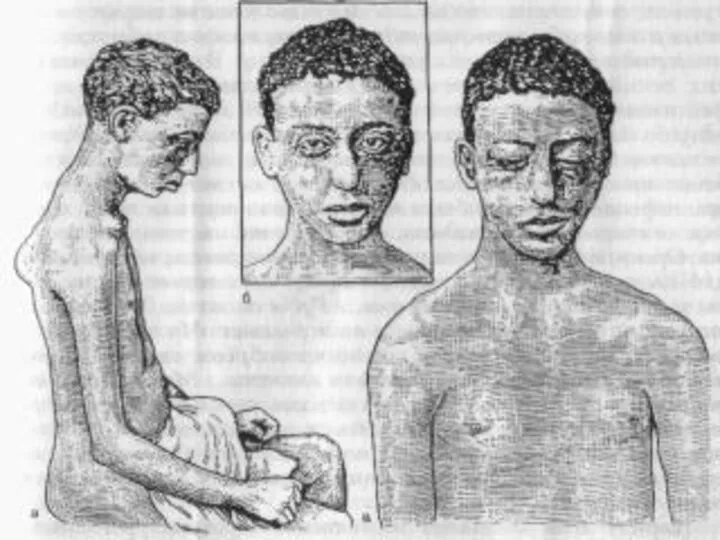

Спинальная амиотрофия Вердига – Гоффмана.
Выделяют формы:
Врожденная форма.
Ранняя детская форма.
Поздняя форма.
Спинальная амиотрофия Вердига – Гоффмана.
Выделяют формы:
Врожденная форма.
Ранняя детская форма.
Поздняя форма.

Врожденная форма:
Начало внутриутробно.
Слабое шевеление плода.
При рождении вялый паралич в проксимальных
Врожденная форма:
Начало внутриутробно.
Слабое шевеление плода.
При рождении вялый паралич в проксимальных

Ранняя детская форма:
Начало до 1,5 лет.
После интеркурретного заболевания ребенок начинает терять
Ранняя детская форма:
Начало до 1,5 лет.
После интеркурретного заболевания ребенок начинает терять

Поздняя форма:
Начало в 1,5 – 2 года.
Вялые параличи проксимальных отделов ног,
Поздняя форма:
Начало в 1,5 – 2 года.
Вялые параличи проксимальных отделов ног,
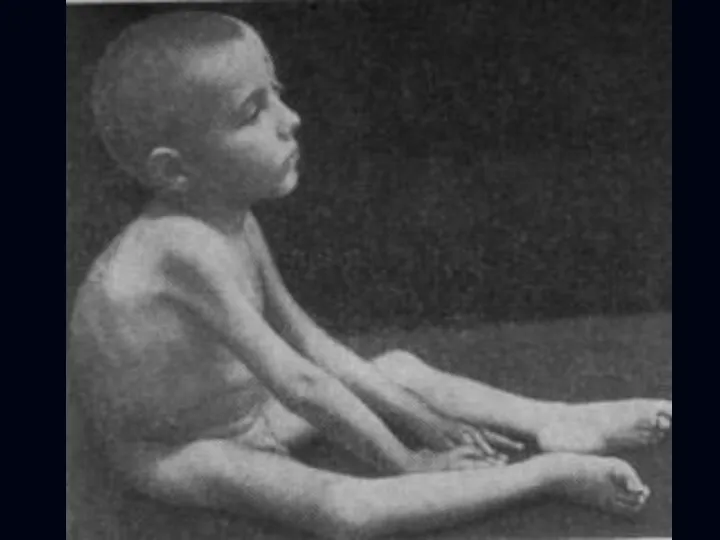
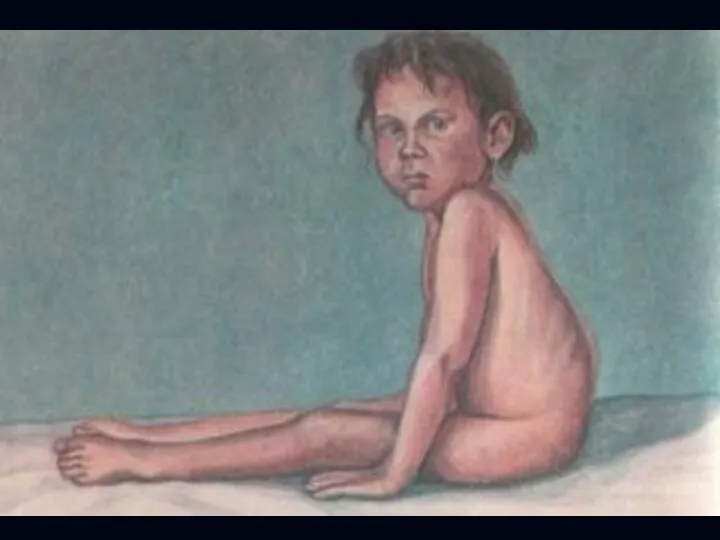
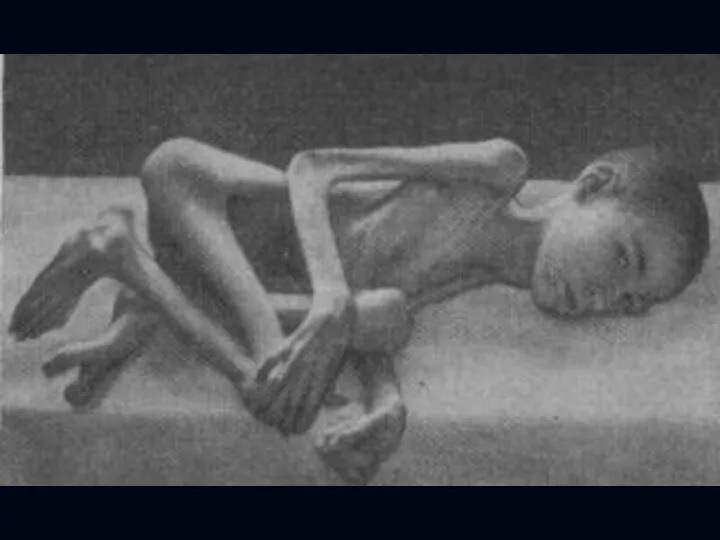

Спинальная амиотрофия Кугельберга-Веландера
(псевдомиопатическая).
Тип наследования аутосомно-рецессивный.
Начало в 6 лет.
Течение доброкачественное, ещё в
Спинальная амиотрофия Кугельберга-Веландера
(псевдомиопатическая).
Тип наследования аутосомно-рецессивный.
Начало в 6 лет.
Течение доброкачественное, ещё в

Невральная амиотрофия Шарко-Мари
Тип наследования аутосомно-доминантный.
Начальные проявления с 13-17 лет.
Характерны дистальные парезы,
Невральная амиотрофия Шарко-Мари
Тип наследования аутосомно-доминантный.
Начальные проявления с 13-17 лет.
Характерны дистальные парезы,

Атрофия мышц голеней и нижней трети бедер.
Ноги по виду напоминают «ноги
Атрофия мышц голеней и нижней трети бедер.
Ноги по виду напоминают «ноги

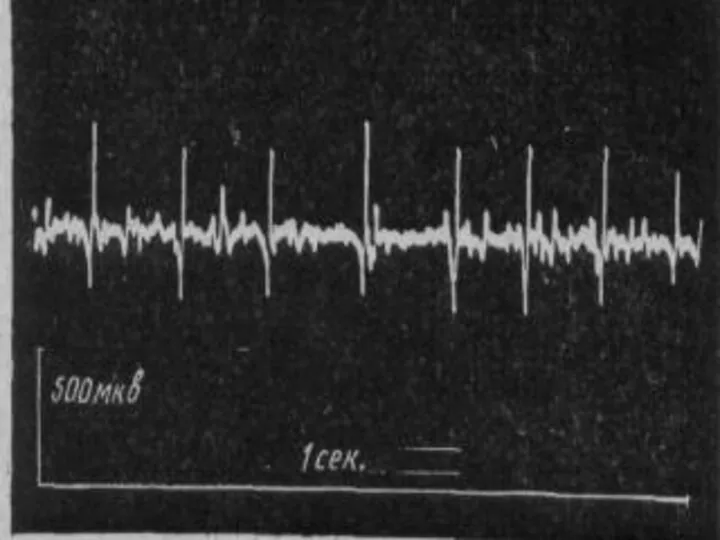
Миастения:
Появляется утомляемость, распространяется на мышцы: губ, жевательные, глазодвигательные, глотательные.
Наблюдается птоз, диплопия,
Миастения:
Появляется утомляемость, распространяется на мышцы: губ, жевательные, глазодвигательные, глотательные.
Наблюдается птоз, диплопия,

Утомляемость появляется в мышцах конечностей.
Наблюдается увеличение вилочковой железы.
Заболевание имеет тенденцию к
Наблюдается увеличение вилочковой железы.
Заболевание имеет тенденцию к

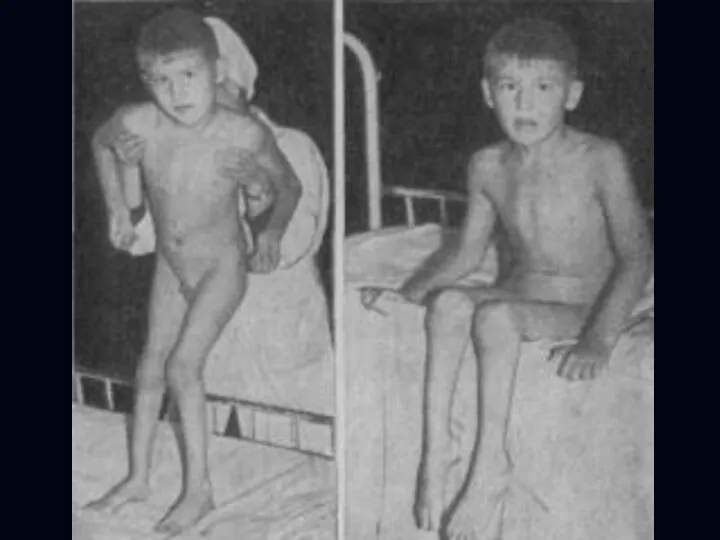
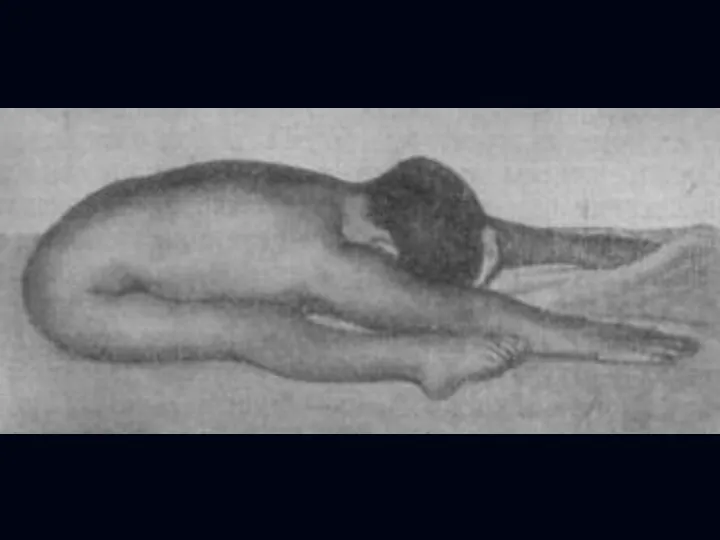
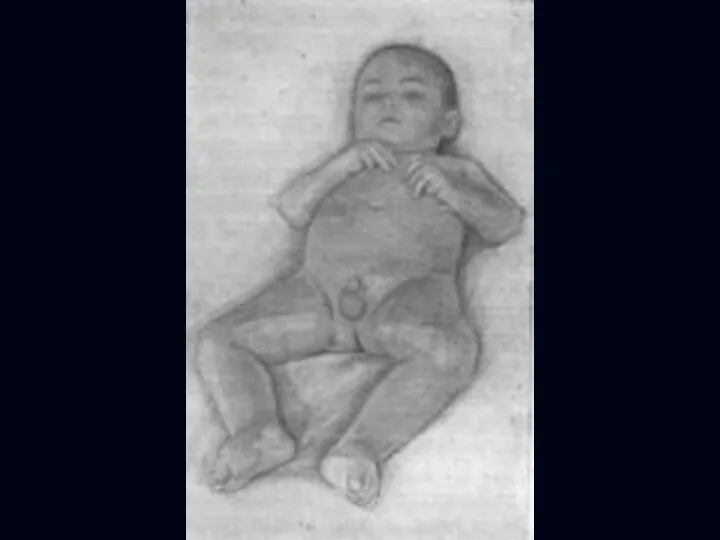
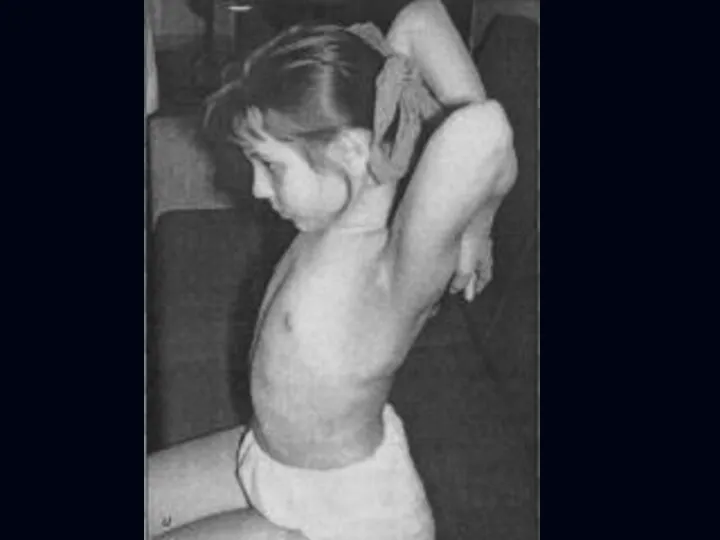
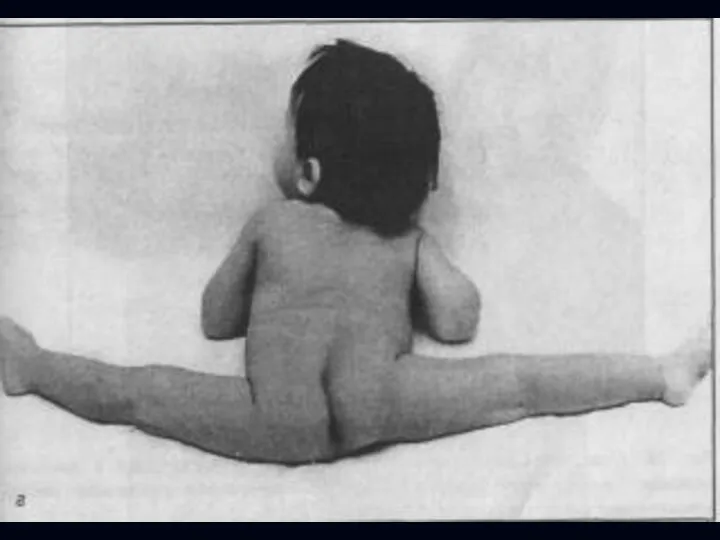
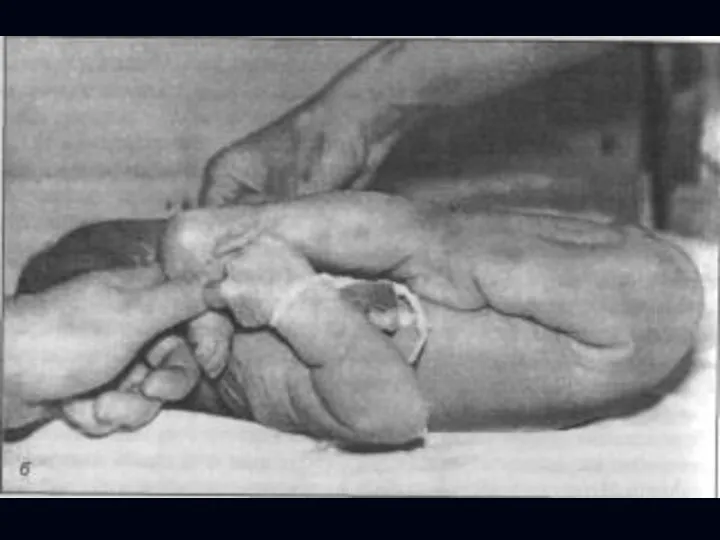
Врожденная миатония Оппенгейма:
С рождения – мало активных движений.
Резкая гипотония или полная
Врожденная миатония Оппенгейма:
С рождения – мало активных движений.
Резкая гипотония или полная

Отсутствуют также гипертрофия и псевдогипертрофия.
Лицевые мышцы поражаются редко.
Психика сохранена.
Тазовых и сенсорных
Отсутствуют также гипертрофия и псевдогипертрофия.
Лицевые мышцы поражаются редко.
Психика сохранена.
Тазовых и сенсорных


Пароксизмальная миоплегия
- генетически детерминированные нервно-мышечные заболевания, обусловленные нарушениями обмена
Пароксизмальная миоплегия
- генетически детерминированные нервно-мышечные заболевания, обусловленные нарушениями обмена

Гипокалиемическая форма:
1. Начало от 10 до 16 лет.
2. Возникновение приступов
Гипокалиемическая форма:
1. Начало от 10 до 16 лет.
2. Возникновение приступов

6. При парциальном приступе мышечная слабость и парез или плегия развивается
6. При парциальном приступе мышечная слабость и парез или плегия развивается

Гиперкалиемическая форма:
1. Начало от 10 до 18 лет.
2. Возникают приступы пароксизмаль-ного
Гиперкалиемическая форма:
1. Начало от 10 до 18 лет.
2. Возникают приступы пароксизмаль-ного

5. В последующем развивается мышечная слабость, гипотония мышц, снижение сухожильных рефлексов.
6.
5. В последующем развивается мышечная слабость, гипотония мышц, снижение сухожильных рефлексов.
6.

Нормокалиемическая форма:
1. Начало в первые 10 лет жизни.
2. Сочетание признаков
Нормокалиемическая форма:
1. Начало в первые 10 лет жизни.
2. Сочетание признаков

Дополнительные методы обследования:
Электромиография (ЭМГ).
Биопсия скелетной мышцы.
Биохимический анализ крови (БАК).
Компьютерная томография скелетных
Дополнительные методы обследования:
Электромиография (ЭМГ).
Биопсия скелетной мышцы.
Биохимический анализ крови (БАК).
Компьютерная томография скелетных

Лечение прогрессирующих мышечных миопатий:
Для улучшения трофики: АТФ.
Антихолинэстеразные препараты: галантамин.
Витаминотерапия:
витамины
Лечение прогрессирующих мышечных миопатий:
Для улучшения трофики: АТФ.
Антихолинэстеразные препараты: галантамин.
Витаминотерапия:
витамины

Лечение прогрессирующих мышечных амиотрофий:
Для улучшения трофики: АТФ, глютаминовая кислота, метионин, лейцин.
Антихолинэстеразные
Лечение прогрессирующих мышечных амиотрофий:
Для улучшения трофики: АТФ, глютаминовая кислота, метионин, лейцин.
Антихолинэстеразные

Лечение невральной амиотрофии Шарко-Мари:
Антихолинэстеразные препараты: прозерин, дибазол.
Для улучшения трофики: АТФ.
ЛФК,
Лечение невральной амиотрофии Шарко-Мари:
Антихолинэстеразные препараты: прозерин, дибазол.
Для улучшения трофики: АТФ.
ЛФК,

Лечение миастении:
Антихолинэстеразные препараты:
прозерин, местинон, оксазил.
Препарат, увеличивающий нервно-мышечную передачу – хлористый
Лечение миастении:
Антихолинэстеразные препараты:
прозерин, местинон, оксазил.
Препарат, увеличивающий нервно-мышечную передачу – хлористый

Лечение пароксизмальной миоплегии:
Во время приступа.
Высокие дозы хлорида калия внутрь (10-15 г
Лечение пароксизмальной миоплегии:
Во время приступа.
Высокие дозы хлорида калия внутрь (10-15 г

В межприступный период:
1) Диета с высоким содержанием калия и ограничением углеводов
В межприступный период:
1) Диета с высоким содержанием калия и ограничением углеводов
 Этиология и патогенез болезней пародонта
Этиология и патогенез болезней пародонта Жүкті әйелдерді амбулаториялық жағдайда жүргізу
Жүкті әйелдерді амбулаториялық жағдайда жүргізу Rak piersi. Leczenie systemowe
Rak piersi. Leczenie systemowe 170 years since the birth of Ivan Pavlov. Traditions and innovations in Cognitive Behavioral Therapy
170 years since the birth of Ivan Pavlov. Traditions and innovations in Cognitive Behavioral Therapy Оқушының дұрыс тамақтануы - жаңа оқу жылындағы жетістіктердің негізі
Оқушының дұрыс тамақтануы - жаңа оқу жылындағы жетістіктердің негізі Рецепт. Структура рецепта
Рецепт. Структура рецепта Особенности проведения медицинских осмотров обучающихся в целях раннего выявления потребления наркотических средств
Особенности проведения медицинских осмотров обучающихся в целях раннего выявления потребления наркотических средств Ротавирусная инфекция
Ротавирусная инфекция Классификация акушерских кровотечений во время беременности
Классификация акушерских кровотечений во время беременности Оказание первой помощи. Современные требования. Часть 1
Оказание первой помощи. Современные требования. Часть 1 Сүйектің сынуы. Буын шығуы соғып алу. Сіңір созылуы
Сүйектің сынуы. Буын шығуы соғып алу. Сіңір созылуы Гостра серцева недостатність
Гостра серцева недостатність Кандидоз. Причины кандидоза
Кандидоз. Причины кандидоза Старение иммунной системы и ее взаимосвязь с течением коронавирусной инфекции (COVID-19)
Старение иммунной системы и ее взаимосвязь с течением коронавирусной инфекции (COVID-19) Современные лекарственные формы и системы доставки лекарственных средств
Современные лекарственные формы и системы доставки лекарственных средств Современные направления комплексного ухода за пациентами с язвенной болезнью 12-ти перстной кишки
Современные направления комплексного ухода за пациентами с язвенной болезнью 12-ти перстной кишки Острая гнойная инфекция костей и суставов
Острая гнойная инфекция костей и суставов Icon. Современный метод лечения кариеса
Icon. Современный метод лечения кариеса Сестринские манипуляции. Постановка очистительной клизмы
Сестринские манипуляции. Постановка очистительной клизмы Анатомия опорно-двигательного аппарата
Анатомия опорно-двигательного аппарата Тамақтану физиологиясы курсының пәні мен міндеттері
Тамақтану физиологиясы курсының пәні мен міндеттері Задержка роста, не связанная с патологией гипофиза
Задержка роста, не связанная с патологией гипофиза Основные виды патологий зрительного нерва
Основные виды патологий зрительного нерва Иммуностимуляторы
Иммуностимуляторы Скульптор среди медиков. Всё о профессии зубного техника
Скульптор среди медиков. Всё о профессии зубного техника Диспансеризация населения
Диспансеризация населения Вскармливание детей первого года жизни
Вскармливание детей первого года жизни Новости nCov-2019 (c 11/02 – COVID-2019, SARS CoV-2)
Новости nCov-2019 (c 11/02 – COVID-2019, SARS CoV-2)