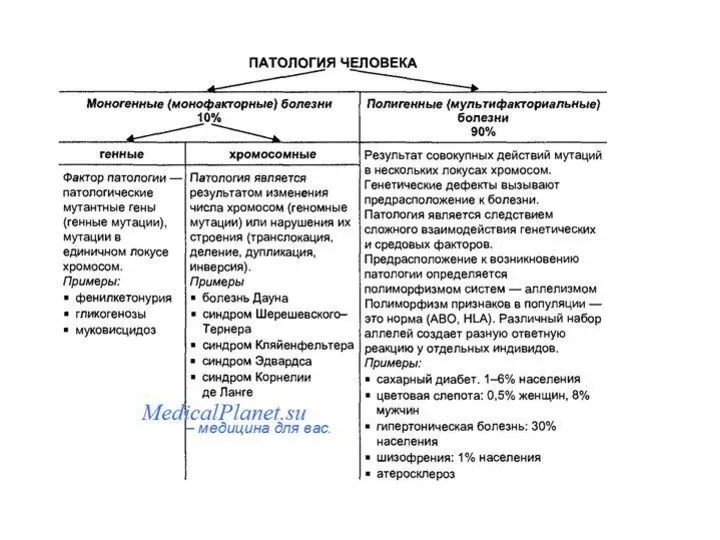
- Наследственность и ее формы. Наследственные болезни как вариант изменчивости

Содержание
- 2. План лекции: Наследственность и среда. Изменчивость и ее формы. Понятие о наследственных болезнях. Методы изучения наследственности
- 3. Преемственность живого на Земле и столь большое разнообразие форм на нашей планете происходит благодаря таким всеобщим
- 4. Наследственность — способность живых организмов передавать от поколения к поколению анатомические, физиологические, биохимические особенности своей организации.
- 5. ИЗМЕНЧИВОСТЬ - ЭТО СПОСОБНОСТЬ ОРГАНИЗМА ПРИОБРЕТАТЬ НОВЫЕ ПРИЗНАКИ В ПРОЦЕССЕ ОНТОГЕНЕЗА
- 6. ИЗМЕНЧИВОСТЬ НАСЛЕДСТВЕННАЯ (ГЕНОТИПИЧЕСКАЯ) НЕНАСЛЕДСТВЕННАЯ(ФЕНОТИПИЧЕСКАЯ) возрастная (онтогенетическая), проявляющаяся в изменении всего комплекса морфофизиологических и биохимических признаков организма
- 7. МОДИФИКАЦИОННАЯ ИЗМЕНЧИВОСТЬ УСЛОВНАЯ КЛАССИФИКАЦИЯ: По изменяющимся признакам организма: морфологические изменения физиологические и биохимические адаптации — гомеостаз
- 8. УСЛОВНАЯ КЛАССИФИКАЦИЯ МОДИФИКАЦИОННОЙ ИЗМЕНЧИВОСТИ (продолжение) По значению: модификации (полезные для организма — проявляются как приспособительная реакция
- 9. ХАРАКТЕРИСТИКА МОДИФИКАЦИОННОЙ ИЗМЕНЧИВОСТИ обратимость — изменения исчезают при смене специфических условий окружающей среды, спровоцировавших их групповой
- 10. НОРМА РЕАКЦИИ - СВОЙСТВО ГЕНОТИПА ОБЕСПЕЧИВАТЬ В ОПРЕДЕЛЕННЫХ ПРЕДЕЛАХ РАЗВИТИЕ ДАННОГО ОНТОГЕНЕЗА В ЗАВИСИМОСТИ ОТ МЕНЯЮЩИХСЯ
- 11. Варианта — это единичное выражение развития признака. Размах вариаций и частоту встречаемости отдельных вариант изучают с
- 12. ИЗМЕНЧИВОСТЬ НАСЛЕДСТВЕННАЯ (ГЕНОТИПИЧЕСКАЯ) НЕНАСЛЕДСТВЕННАЯ(ФЕНОТИПИЧЕСКАЯ) возрастная (онтогенетическая), проявляющаяся в изменении всего комплекса морфофизиологических и биохимических признаков организма
- 13. Комбинативная изменчивость – возникновение новых комбинаций генов в генотипе. Каждая новая комбинация при половом размножении приводит
- 14. Комбинативная изменчивость связана с появлением новых соединений генов в генотипе: независимое расхождение хромосом в мейозе; рекомбинации
- 15. Мутация - любое долговременное изменение в последовательности ДНК. источник генетического разнообразия. мутация возникает внезапно, скачкообразно; мутации
- 16. Факторы, способные вызывать мутации, называют мутагенами. Виды мутагенов: физические (различные виды излучений, температура); химические (формалин, бензпирен,
- 17. Мутации можно подразделить: По проявлению мутации в гетерозиготе: Доминантные Рецессивные По причине возникновения: Спонтанные Индуцированные По
- 18. По изменению фенотипа (по Мёллеру): аморфные мутации (греч. «а» – отрицание, «морфа»-форма) неактивны в отношении типичного
- 19. По значению в эволюционном процессе Прогрессивные Нейтральные Ретрогрессивные По продолжительности жизни Полулетальные Летальные По степени вовлеченности
- 21. Геномные мутации - изменение числа хромосом Полиплоидия - кратное увеличение гаплоидного набора хромосом (Зп, 4п, и
- 22. Полиплоидия
- 24. Нарушения числа аутосом Синдром Патау (трисомия по 13 -и паре хромосом)
- 25. При синдроме Патау наблюдаются тяжелые врождённые пороки. Дети с синдромом Патау рождаются с массой тела ниже
- 26. Синдром Эдвардса (трисомия 18-й пары хромосом)
- 27. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет
- 29. Синдром Дауна (трисомия по 21 - и паре хромосом)
- 30. Обычно синдрому Дауна сопутствуют следующие внешние признаки (согласно данным из брошюры центра «Даунсайд Ап»): «плоское лицо»
- 31. клинодактилия 5-го пальца (искривлённый мизинец) — 60 % аркообразное нёбо — 58 % плоская переносица —
- 32. Нарушения структуры хромосом хромосомные аберрации
- 33. (дефишенси) Реципрокные взаимный обмен участками между хромосомами Робертсоновские или центрические слияния, при которых происходит слияние акроцентрических
- 34. Схема образования робертсоновской транслокации (а), изохромосом (б) и кольцевой хромосомы (в), A и В — плечи
- 35. ОРБЕЛИ СИНДРОМ - Описан в 1962 г. Н. Wang. синдром хромосомы 13q-) – хромосомное заболевание, обусловленное
- 36. Популяционная частота неизвестна. Соотношение полов — M1:Ж1.
- 37. Клинические проявления: дефицит массы тела, возникающий уже пренатально; постнатальная задержка роста; микроцефалия; преждевременное зарастание черепных швов;
- 38. Фенотипы детей с синдромом 13q- (Синдром Орбели) Kirchhoff M. et al.Phenotype and 244k Array-CGH Characterization of
- 39. Делеция (с утратой от трети до половины, реже полная утрата) короткого плеча пятой хромосомы Синдром «
- 40. ТРАНСЛОКАЦИИ хромосома №2 человека образована при транслокации, произошедшей у наших предков после ответвления от ствола всех
- 41. Реципрокная транслокация фрагментов между хромосомами 8 и 14 в лимфоцитах человека приводит к лимфоме Бёркита: к
- 42. Транслокация (перемещение) хромосомы 21 пары на 15 пару транслокационный синдром Дауна Примерно 4% больных СД имеют
- 43. Основная часть транслокаций, обусловливающих синдром Дауна, относится к центральному соединению 21-й и D-хромосом; примерно половина из
- 44. ГЕННЫЕ МУТАЦИИ В результате генных мутаций происходят: Замены, Делеции Вставки одного или нескольких нуклеотидов, Транслокации, Дупликации
- 45. Генные дупликации — удвоение пары или нескольких пар нуклеотидов (удвоение пары Г—Ц). Всякий раз, когда несколько
- 46. Дупликации, инсерции и делеции могут приводить к изменению рамки считывания генетического кода.
- 47. Генные инверсии — перестановка фрагмента гена (во фрагменте исходная последовательность нуклеотидов Т—А, Г—Ц за меняется на
- 48. Рекомбинации при кроссинговере Неравный кроссинговер между генами для красного и зеленого цветового зрения Законная рекомбинация (обмен
- 49. 1. Missense-мутация. 2. Мутация со сдвигом рамки. 3. Nonsense-мутация. 4. Синонимическая missence-мутация. ТОЧКОВЫЕ МУТАЦИИ
- 50. 1. Missense-мутация. В одном из триплетов происходит замена одного основания (например, ЦТТ→ГТТ), в результате чего измененный
- 51. 3. Nonsense-мутация. В результате замены одного основания возникает новый триплет, представляющий собой терминирующий кодон. В генетическом
- 52. Один из наиболее частых типов мутаций замена пары нуклеотидов Т—А на Ц—Г при этом общее число
- 54. Генные болезни Серповидноклеточная анемия Фенилкетонурия Галактоземия Альбинизм Нарушение цветового зрения
- 55. Серповидноклеточная анемия
- 56. Фенилкетонурия
- 58. Типичные клинические признаки: грубый дефект лицевой части черепа, косоглазие, колобомы век; размер рта, подбородка и ушей
- 59. Эта мутация влияет на развитие костной и других тканей лица. Большинство пострадавших имеют неразвитые лицевые кости,
- 60. ИЗУЧЕНИЕ ГЕНЕТИКИ ЧЕЛОВЕКА
- 61. Изучение генетики человека связано с рядом особенностей и объективных трудностей: 1) сложный кариотип; 2) позднее половое
- 62. Клиникогенеалогический метод был предложен в 1883 г. Ф. Гальтоном. Он основан на построении родословных и прослеживании
- 63. Примеры родословных человека: а — с рецессивными признаками; б — по полидактилии — шестипалость (доминантный признак)
- 64. На рисунке представлена родословная семьи с хромосомными аберрациями. Среди родственников мужа - умственно отсталая сестра, а
- 65. Родословная с Х-сцепленной рецессивной гемофилией В в европейских королевских домах. Королева Виктория (1.2) была гетерозиготой. Она
- 66. 1. Сбор сведений у пробанда (лицо, к которому строится родословная) о наличии или отсутствии анализируемого признака
- 67. Близнецовый метод изучения генетики человека введен в медицинскую практику Ф. Гальтоном в 1876 г. Позволяет определить
- 68. Процент сходства близнецов по изучаемому признаку называется конкордантностью, а процент различия - дискордантностью. Поскольку монозиготные близнецы
- 69. Для оценки роли наследственности и среды в развитии того или иного признака используется формула Хольцингера: Если
- 70. Пренатальная диагностика врожденных и наследственных болезней - это комплексная отрасль медицины
- 71. При организации и развитии системы пренатальной диагностики должны выполняться следующие условия: - Диагностические процедуры должны быть
- 72. Пренатальная диагностика должна включать два этапа: - первый этап - выявление женщин (точнее, семей) с повышенным
- 73. Показания к проведению пренатальной диагностики: 1. Возраст матери 35 лет; 2. Наличие в семье предыдущего ребенка
- 74. Методы пренатальной диагностики: ультразвуковая диагностика (УЗИ), оперативная техника (хорионбиопсию, амнио-и кордоцентез, биопсию мышц и кожи плода),
- 75. Амниоцентез Биохимическое исследование околоплодных вод на выявление маркеров генетического заболевания Кариотипирование клеток плода – выявление геномного
- 76. ЦИТОГЕНЕТИЧЕСКИЕ МЕТОДЫ
- 77. Цитологические методы связаны с проведением окрашивания цитологического материала и последующей микроскопией. В эту группу методов входят:
- 78. Определение тельца Бара и f-тельца
- 79. Метод кариотипирования
- 80. Биохимические методы. Позволяют выявить либо аномальные белки-ферменты, либо промежуточные продукты обмена, свидетельствующие о наличии болезни. Сегодня
- 82. ДНК-диагностика Это наиболее точный метод диагностики моногенных наследственных заболеваний. Преимущества метода: 1) позволяет определить причину заболевания
- 83. АКСИОМЫ МЕДЕЦИНСКОЙ ГЕНЕТИКИ Наследственные болезни являются общей частью наследственной изменчивости человека. В развитии наследственных признаков или
- 84. Ответить на вопросы: Какие причины могут лежать в основе синдрома Дауна? Что такое Робертсоновские мутации? Что
- 86. Скачать презентацию

План лекции:
Наследственность и среда.
Изменчивость и ее формы.
Понятие о наследственных
План лекции:
Наследственность и среда.
Изменчивость и ее формы.
Понятие о наследственных

Преемственность живого на Земле и столь большое разнообразие форм на нашей
Преемственность живого на Земле и столь большое разнообразие форм на нашей

Наследственность — способность живых организмов передавать от поколения к поколению анатомические, физиологические,
Наследственность — способность живых организмов передавать от поколения к поколению анатомические, физиологические,

ИЗМЕНЧИВОСТЬ - ЭТО СПОСОБНОСТЬ ОРГАНИЗМА ПРИОБРЕТАТЬ НОВЫЕ ПРИЗНАКИ В ПРОЦЕССЕ ОНТОГЕНЕЗА
ИЗМЕНЧИВОСТЬ - ЭТО СПОСОБНОСТЬ ОРГАНИЗМА ПРИОБРЕТАТЬ НОВЫЕ ПРИЗНАКИ В ПРОЦЕССЕ ОНТОГЕНЕЗА
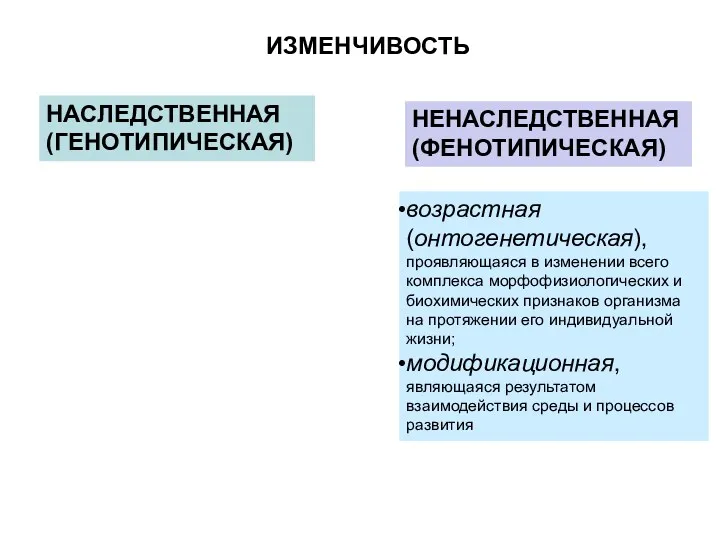
ИЗМЕНЧИВОСТЬ
НАСЛЕДСТВЕННАЯ (ГЕНОТИПИЧЕСКАЯ)
НЕНАСЛЕДСТВЕННАЯ(ФЕНОТИПИЧЕСКАЯ)
возрастная (онтогенетическая), проявляющаяся в изменении всего комплекса морфофизиологических и биохимических
ИЗМЕНЧИВОСТЬ
НАСЛЕДСТВЕННАЯ (ГЕНОТИПИЧЕСКАЯ)
НЕНАСЛЕДСТВЕННАЯ(ФЕНОТИПИЧЕСКАЯ)
возрастная (онтогенетическая), проявляющаяся в изменении всего комплекса морфофизиологических и биохимических

МОДИФИКАЦИОННАЯ ИЗМЕНЧИВОСТЬ
УСЛОВНАЯ КЛАССИФИКАЦИЯ:
По изменяющимся признакам организма:
морфологические изменения
физиологические и биохимические
МОДИФИКАЦИОННАЯ ИЗМЕНЧИВОСТЬ
УСЛОВНАЯ КЛАССИФИКАЦИЯ:
По изменяющимся признакам организма:
морфологические изменения
физиологические и биохимические

УСЛОВНАЯ КЛАССИФИКАЦИЯ МОДИФИКАЦИОННОЙ ИЗМЕНЧИВОСТИ (продолжение)
По значению:
модификации (полезные для организма — проявляются
УСЛОВНАЯ КЛАССИФИКАЦИЯ МОДИФИКАЦИОННОЙ ИЗМЕНЧИВОСТИ (продолжение)
По значению:
модификации (полезные для организма — проявляются

ХАРАКТЕРИСТИКА МОДИФИКАЦИОННОЙ ИЗМЕНЧИВОСТИ
обратимость — изменения исчезают при смене специфических условий окружающей среды,
ХАРАКТЕРИСТИКА МОДИФИКАЦИОННОЙ ИЗМЕНЧИВОСТИ
обратимость — изменения исчезают при смене специфических условий окружающей среды,

НОРМА РЕАКЦИИ - СВОЙСТВО ГЕНОТИПА ОБЕСПЕЧИВАТЬ В ОПРЕДЕЛЕННЫХ ПРЕДЕЛАХ РАЗВИТИЕ ДАННОГО
НОРМА РЕАКЦИИ - СВОЙСТВО ГЕНОТИПА ОБЕСПЕЧИВАТЬ В ОПРЕДЕЛЕННЫХ ПРЕДЕЛАХ РАЗВИТИЕ ДАННОГО
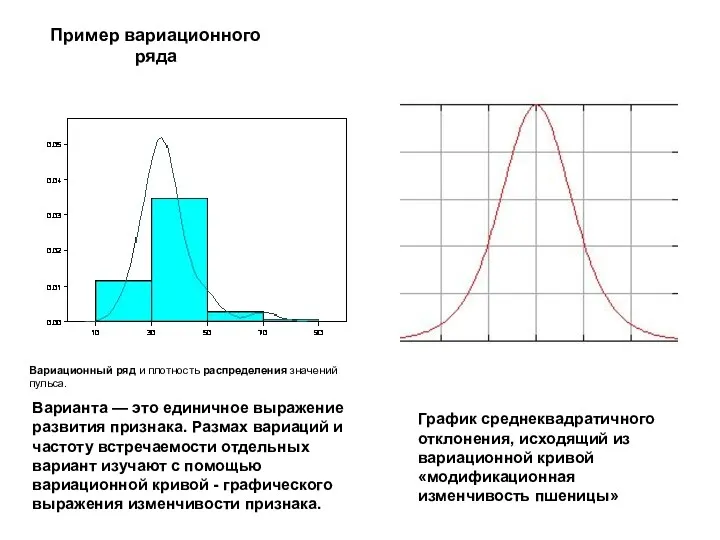
Варианта — это единичное выражение развития признака. Размах вариаций и частоту
Варианта — это единичное выражение развития признака. Размах вариаций и частоту
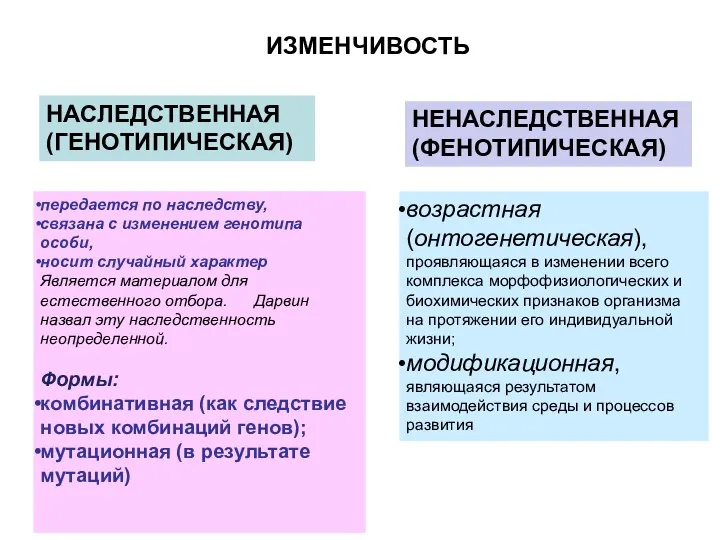
ИЗМЕНЧИВОСТЬ
НАСЛЕДСТВЕННАЯ (ГЕНОТИПИЧЕСКАЯ)
НЕНАСЛЕДСТВЕННАЯ(ФЕНОТИПИЧЕСКАЯ)
возрастная (онтогенетическая), проявляющаяся в изменении всего комплекса морфофизиологических и биохимических
ИЗМЕНЧИВОСТЬ
НАСЛЕДСТВЕННАЯ (ГЕНОТИПИЧЕСКАЯ)
НЕНАСЛЕДСТВЕННАЯ(ФЕНОТИПИЧЕСКАЯ)
возрастная (онтогенетическая), проявляющаяся в изменении всего комплекса морфофизиологических и биохимических

Комбинативная изменчивость – возникновение новых комбинаций генов в генотипе. Каждая новая
Комбинативная изменчивость – возникновение новых комбинаций генов в генотипе. Каждая новая

Комбинативная изменчивость связана с появлением новых соединений генов в генотипе:
независимое расхождение
Комбинативная изменчивость связана с появлением новых соединений генов в генотипе:
независимое расхождение

Мутация - любое долговременное изменение в последовательности ДНК.
источник генетического разнообразия.
мутация возникает
Мутация - любое долговременное изменение в последовательности ДНК.
источник генетического разнообразия.
мутация возникает

Факторы, способные вызывать мутации, называют мутагенами.
Виды мутагенов:
физические (различные виды излучений, температура);
химические
Факторы, способные вызывать мутации, называют мутагенами.
Виды мутагенов:
физические (различные виды излучений, температура);
химические

Мутации можно подразделить:
По проявлению мутации в гетерозиготе:
Доминантные
Рецессивные
По причине возникновения:
Спонтанные
Индуцированные
По уклонению от
Мутации можно подразделить:
По проявлению мутации в гетерозиготе:
Доминантные
Рецессивные
По причине возникновения:
Спонтанные
Индуцированные
По уклонению от

По изменению фенотипа (по Мёллеру):
аморфные мутации (греч. «а» – отрицание, «морфа»-форма)
По изменению фенотипа (по Мёллеру):
аморфные мутации (греч. «а» – отрицание, «морфа»-форма)

По значению в эволюционном процессе
Прогрессивные
Нейтральные
Ретрогрессивные
По продолжительности жизни
Полулетальные
Летальные
По степени вовлеченности генома:
геномные,
хромосомные
генные.
По значению в эволюционном процессе
Прогрессивные
Нейтральные
Ретрогрессивные
По продолжительности жизни
Полулетальные
Летальные
По степени вовлеченности генома:
геномные,
хромосомные
генные.
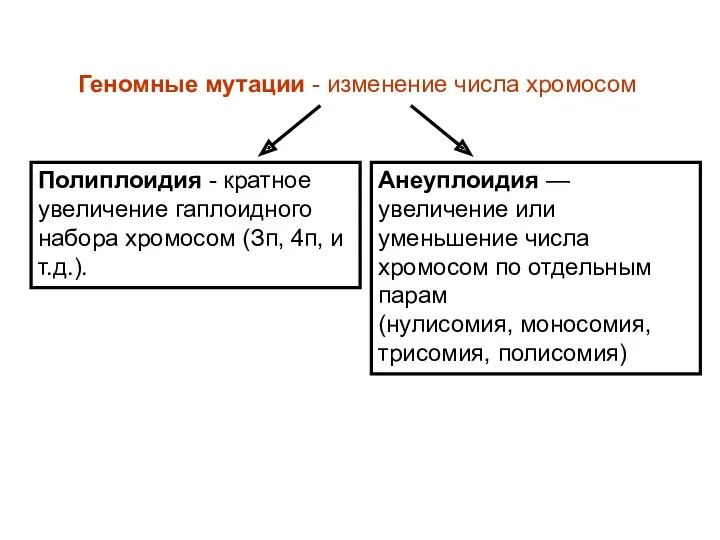
Геномные мутации - изменение числа хромосом
Полиплоидия - кратное увеличение гаплоидного
Геномные мутации - изменение числа хромосом
Полиплоидия - кратное увеличение гаплоидного
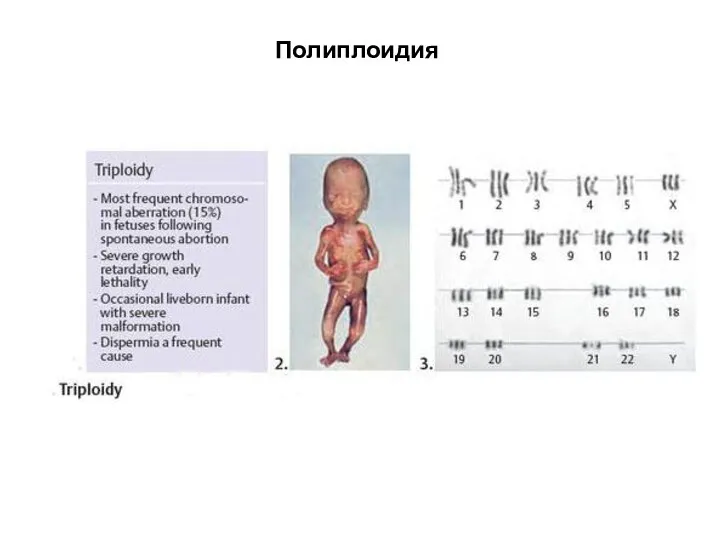
Полиплоидия
Полиплоидия

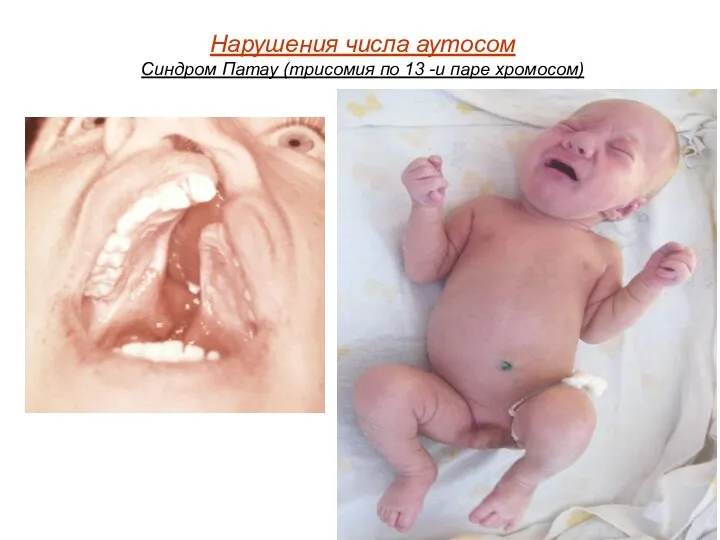
Нарушения числа аутосом
Синдром Патау (трисомия по 13 -и паре хромосом)
Нарушения числа аутосом
Синдром Патау (трисомия по 13 -и паре хромосом)

При синдроме Патау наблюдаются тяжелые врождённые пороки. Дети с синдромом Патау
При синдроме Патау наблюдаются тяжелые врождённые пороки. Дети с синдромом Патау
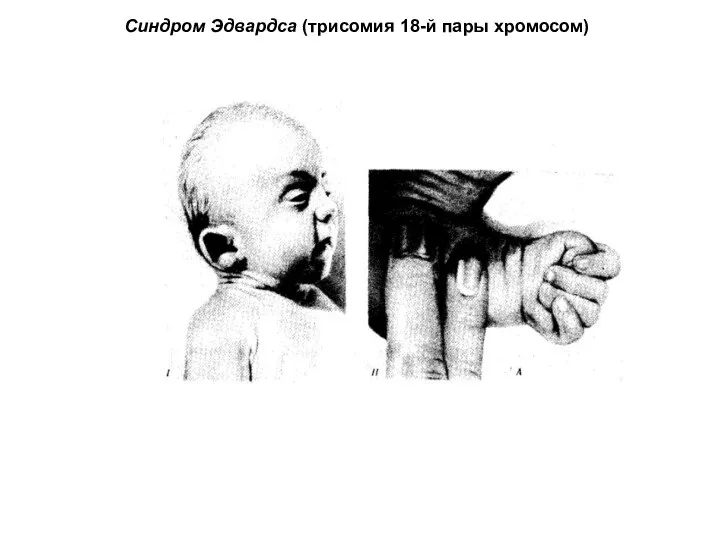
Синдром Эдвардса (трисомия 18-й пары хромосом)
Синдром Эдвардса (трисомия 18-й пары хромосом)

Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и
Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и

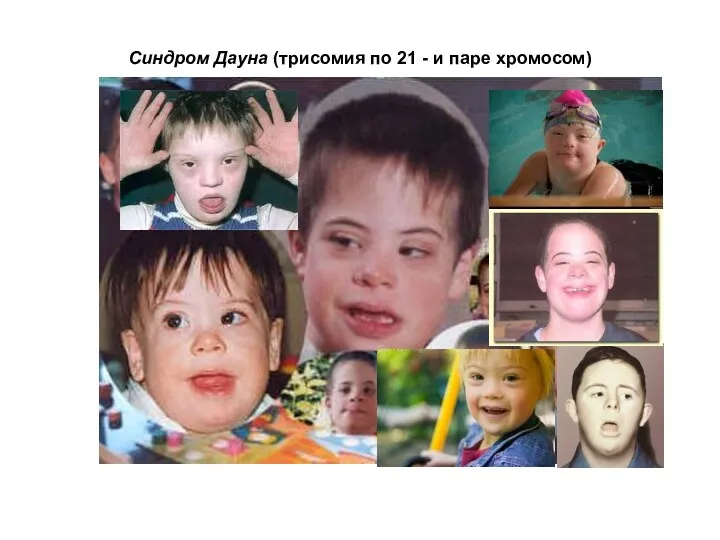
Синдром Дауна (трисомия по 21 - и паре хромосом)
Синдром Дауна (трисомия по 21 - и паре хромосом)

Обычно синдрому Дауна сопутствуют следующие внешние признаки (согласно данным из брошюры
Обычно синдрому Дауна сопутствуют следующие внешние признаки (согласно данным из брошюры

клинодактилия 5-го пальца (искривлённый мизинец) — 60 %
аркообразное нёбо — 58 %
плоская переносица — 52 %
бороздчатый язык — 50 %
поперечная ладонная складка (называемая
клинодактилия 5-го пальца (искривлённый мизинец) — 60 %
аркообразное нёбо — 58 %
плоская переносица — 52 %
бороздчатый язык — 50 %
поперечная ладонная складка (называемая

Нарушения структуры хромосом
хромосомные аберрации
Нарушения структуры хромосом
хромосомные аберрации
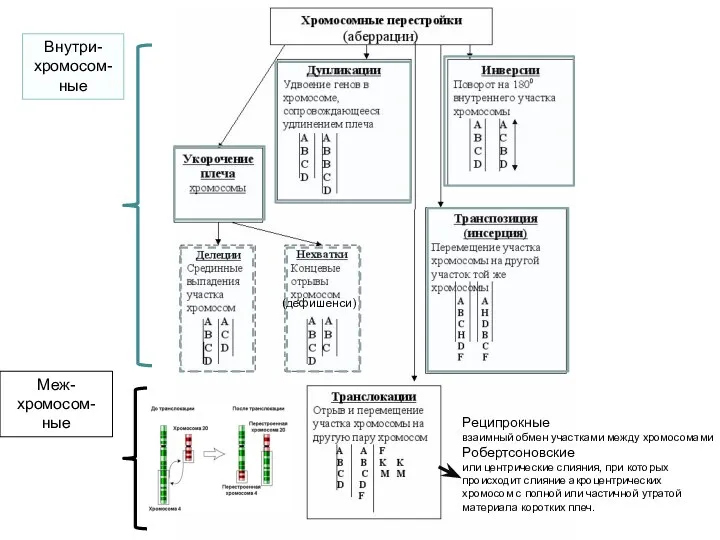
(дефишенси)
Реципрокные
взаимный обмен участками между хромосомами
Робертсоновские
или центрические слияния, при которых происходит слияние
(дефишенси)
Реципрокные
взаимный обмен участками между хромосомами
Робертсоновские
или центрические слияния, при которых происходит слияние
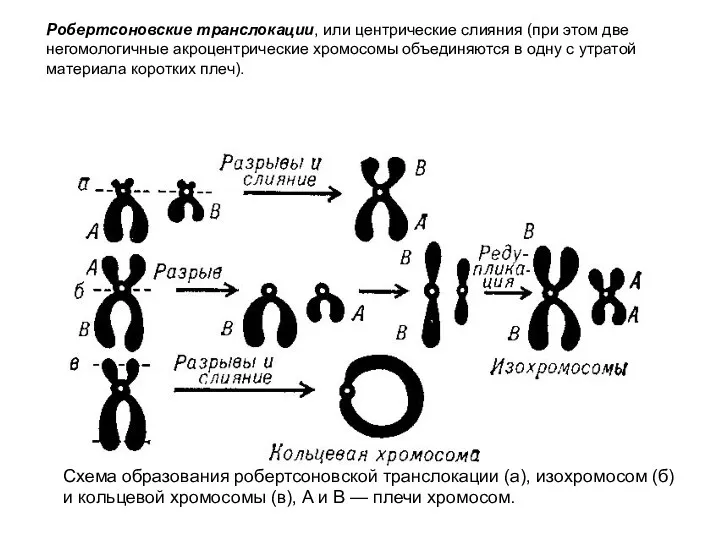
Схема образования робертсоновской транслокации (а), изохромосом (б) и кольцевой хромосомы (в),
Схема образования робертсоновской транслокации (а), изохромосом (б) и кольцевой хромосомы (в),
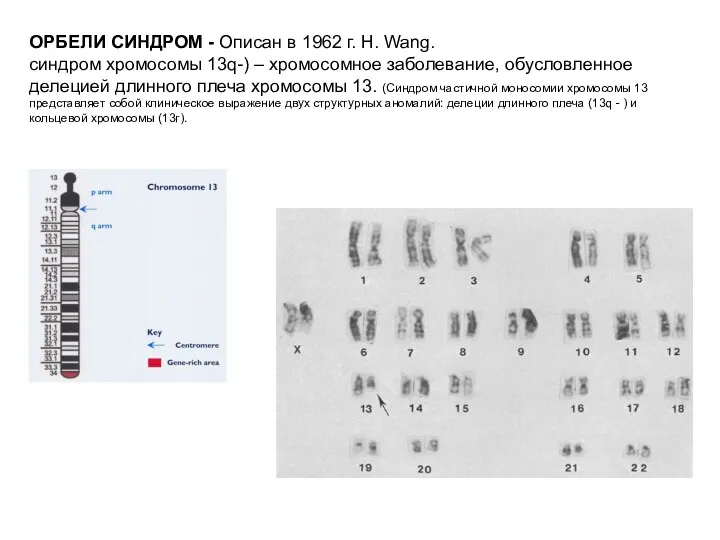
ОРБЕЛИ СИНДРОМ - Описан в 1962 г. Н. Wang.
синдром хромосомы 13q-)
ОРБЕЛИ СИНДРОМ - Описан в 1962 г. Н. Wang.
синдром хромосомы 13q-)
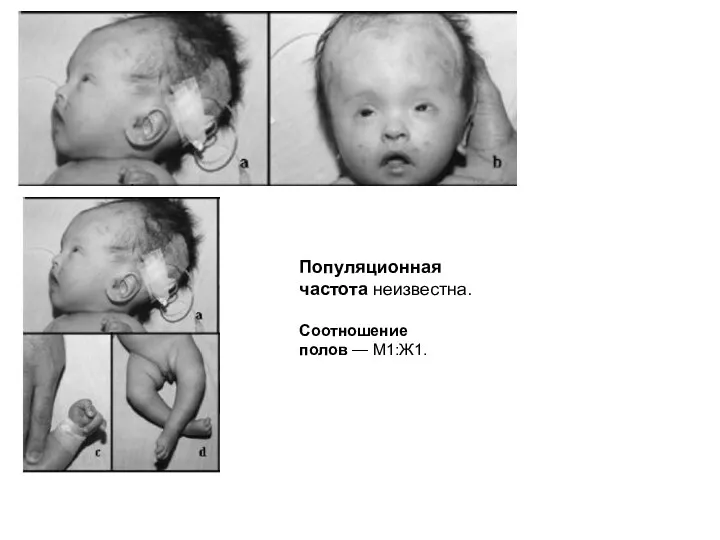
Популяционная частота неизвестна.
Соотношение полов — M1:Ж1.
Популяционная частота неизвестна.
Соотношение полов — M1:Ж1.

Клинические проявления:
дефицит массы тела, возникающий уже пренатально;
постнатальная задержка роста;
Клинические проявления:
дефицит массы тела, возникающий уже пренатально;
постнатальная задержка роста;
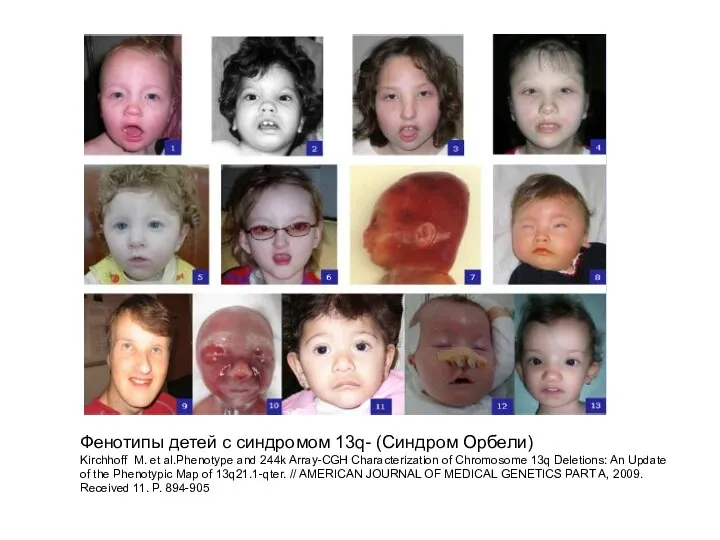
Фенотипы детей с синдромом 13q- (Синдром Орбели)
Kirchhoff M. et al.Phenotype and
Фенотипы детей с синдромом 13q- (Синдром Орбели)
Kirchhoff M. et al.Phenotype and

Делеция (с утратой от трети до половины, реже полная утрата) короткого
Делеция (с утратой от трети до половины, реже полная утрата) короткого
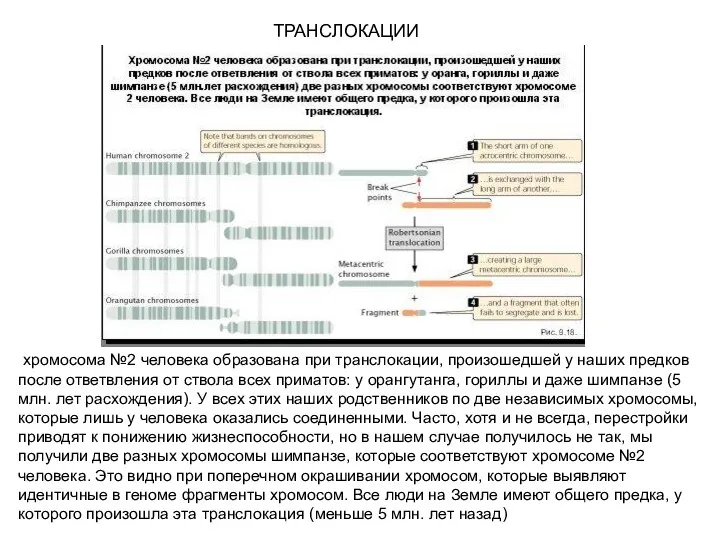
ТРАНСЛОКАЦИИ
хромосома №2 человека образована при транслокации, произошедшей у наших предков после
ТРАНСЛОКАЦИИ
хромосома №2 человека образована при транслокации, произошедшей у наших предков после
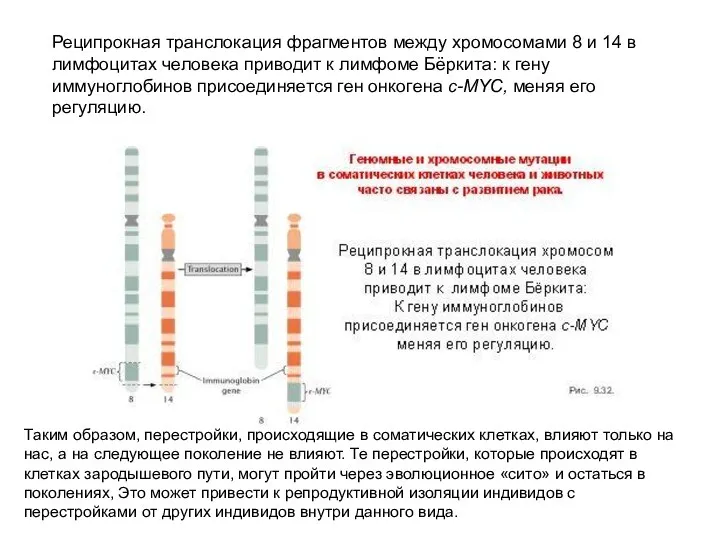
Реципрокная транслокация фрагментов между хромосомами 8 и 14 в лимфоцитах человека
Реципрокная транслокация фрагментов между хромосомами 8 и 14 в лимфоцитах человека

Транслокация (перемещение) хромосомы 21 пары на 15 пару транслокационный синдром Дауна
Транслокация (перемещение) хромосомы 21 пары на 15 пару транслокационный синдром Дауна

Основная часть транслокаций, обусловливающих синдром Дауна, относится к центральному соединению 21-й
Основная часть транслокаций, обусловливающих синдром Дауна, относится к центральному соединению 21-й

ГЕННЫЕ МУТАЦИИ
В результате генных мутаций происходят:
Замены,
Делеции
Вставки одного или нескольких
ГЕННЫЕ МУТАЦИИ
В результате генных мутаций происходят:
Замены,
Делеции
Вставки одного или нескольких

Генные дупликации — удвоение пары или нескольких пар
нуклеотидов (удвоение пары Г—Ц).
Всякий
Генные дупликации — удвоение пары или нескольких пар
нуклеотидов (удвоение пары Г—Ц).
Всякий

Дупликации, инсерции и делеции могут приводить к изменению рамки считывания генетического
Дупликации, инсерции и делеции могут приводить к изменению рамки считывания генетического
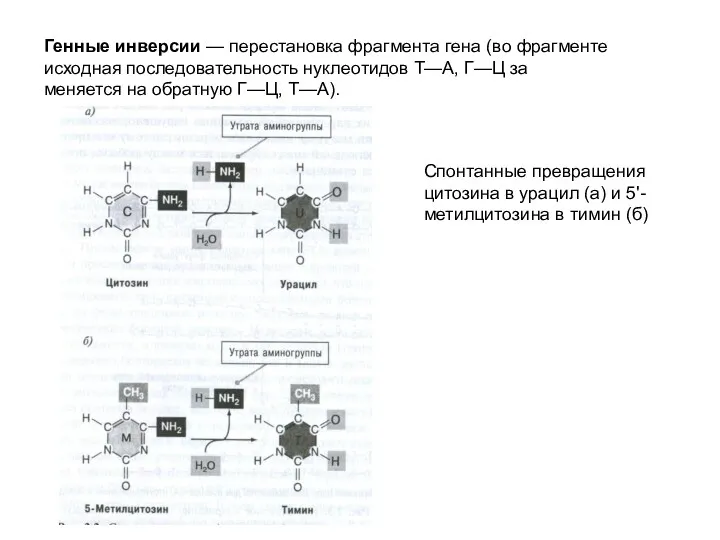
Генные инверсии — перестановка фрагмента гена (во фрагменте исходная последовательность нуклеотидов
Генные инверсии — перестановка фрагмента гена (во фрагменте исходная последовательность нуклеотидов
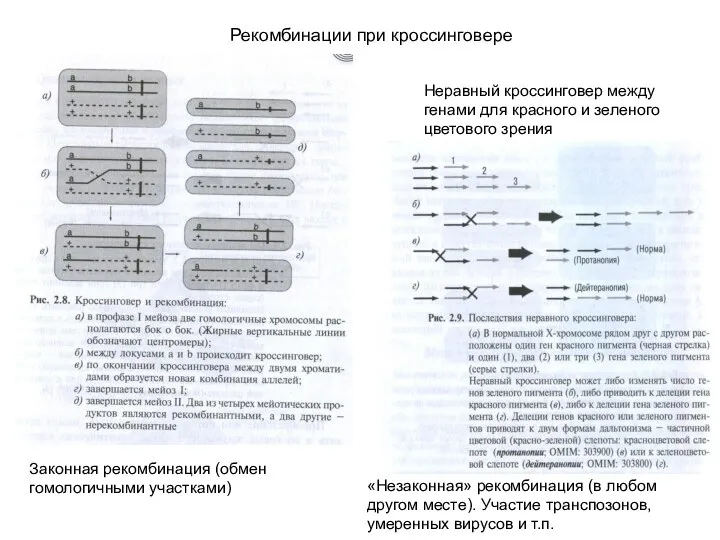
Рекомбинации при кроссинговере
Неравный кроссинговер между генами для красного и зеленого цветового
Рекомбинации при кроссинговере
Неравный кроссинговер между генами для красного и зеленого цветового

1. Missense-мутация.
2. Мутация со сдвигом рамки.
3. Nonsense-мутация.
4. Синонимическая
1. Missense-мутация.
2. Мутация со сдвигом рамки.
3. Nonsense-мутация.
4. Синонимическая

1. Missense-мутация. В одном из триплетов происходит замена одного основания (например,
1. Missense-мутация. В одном из триплетов происходит замена одного основания (например,

3. Nonsense-мутация. В результате замены одного основания возникает новый триплет, представляющий
3. Nonsense-мутация. В результате замены одного основания возникает новый триплет, представляющий
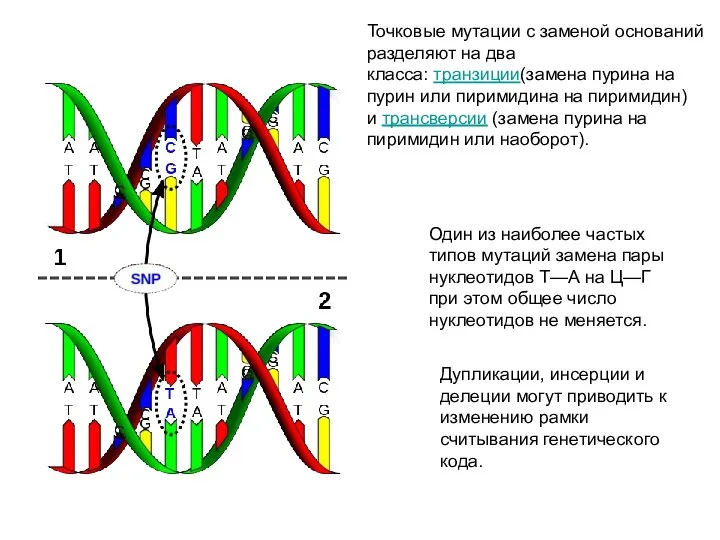
Один из наиболее частых типов мутаций замена пары нуклеотидов Т—А на
Один из наиболее частых типов мутаций замена пары нуклеотидов Т—А на


Генные болезни
Серповидноклеточная анемия
Фенилкетонурия
Галактоземия
Альбинизм
Нарушение цветового зрения
Генные болезни
Серповидноклеточная анемия
Фенилкетонурия
Галактоземия
Альбинизм
Нарушение цветового зрения
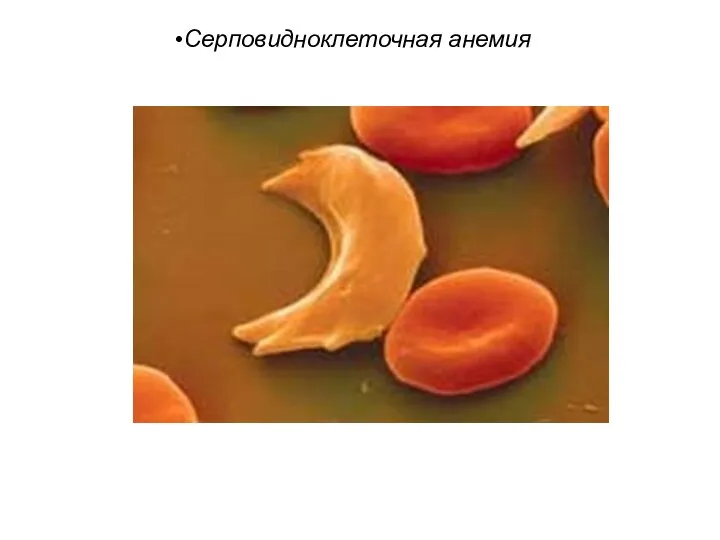
Серповидноклеточная анемия
Серповидноклеточная анемия
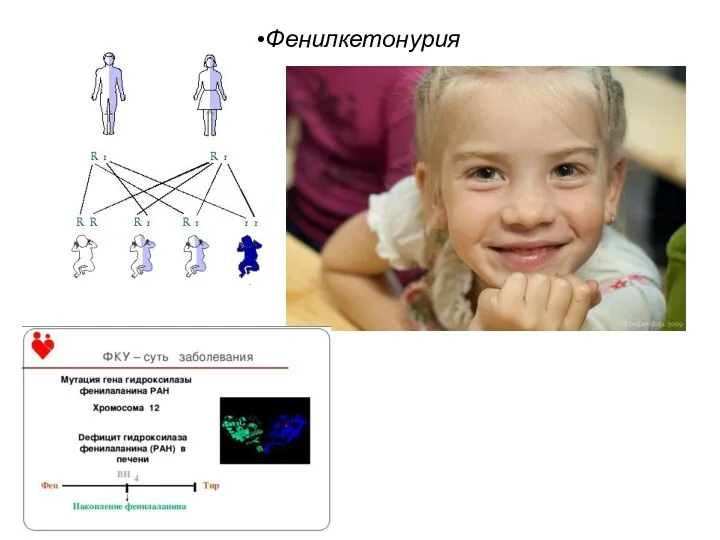
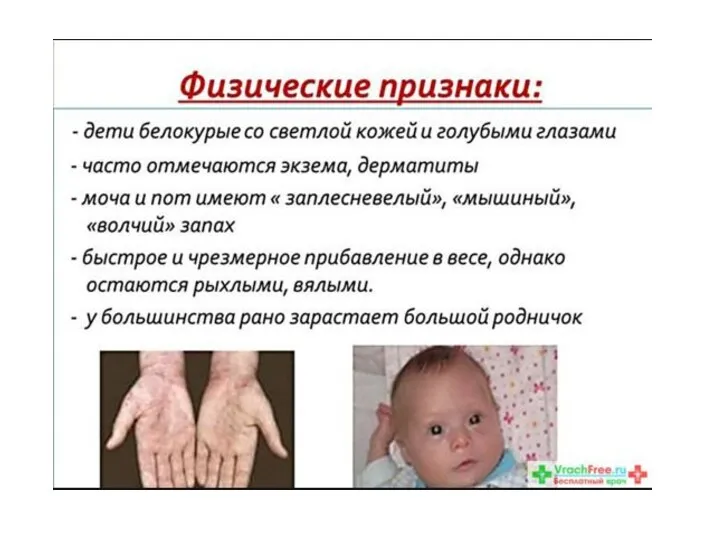
Фенилкетонурия
Фенилкетонурия
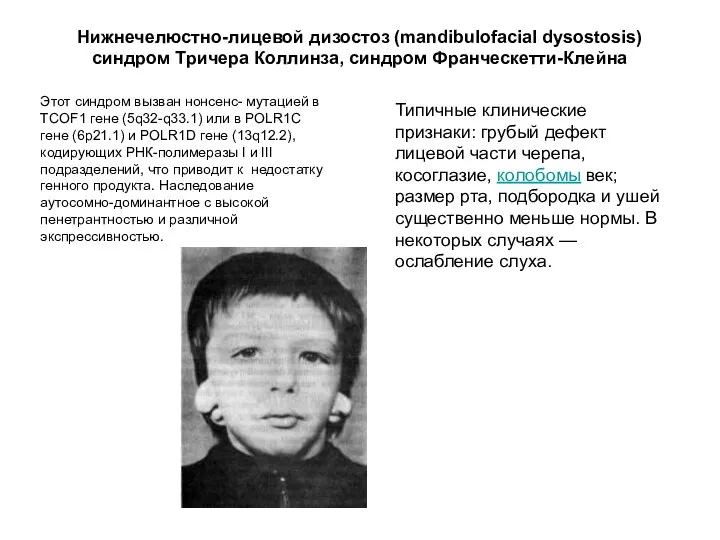
Типичные клинические признаки: грубый дефект лицевой части черепа, косоглазие, колобомы век; размер рта,
Типичные клинические признаки: грубый дефект лицевой части черепа, косоглазие, колобомы век; размер рта,

Эта мутация влияет на развитие костной и других тканей лица. Большинство
Эта мутация влияет на развитие костной и других тканей лица. Большинство

ИЗУЧЕНИЕ ГЕНЕТИКИ ЧЕЛОВЕКА
ИЗУЧЕНИЕ ГЕНЕТИКИ ЧЕЛОВЕКА

Изучение генетики человека связано с рядом особенностей и объективных трудностей:
1)
Изучение генетики человека связано с рядом особенностей и объективных трудностей:
1)

Клиникогенеалогический метод был предложен в 1883 г. Ф. Гальтоном. Он основан
Клиникогенеалогический метод был предложен в 1883 г. Ф. Гальтоном. Он основан
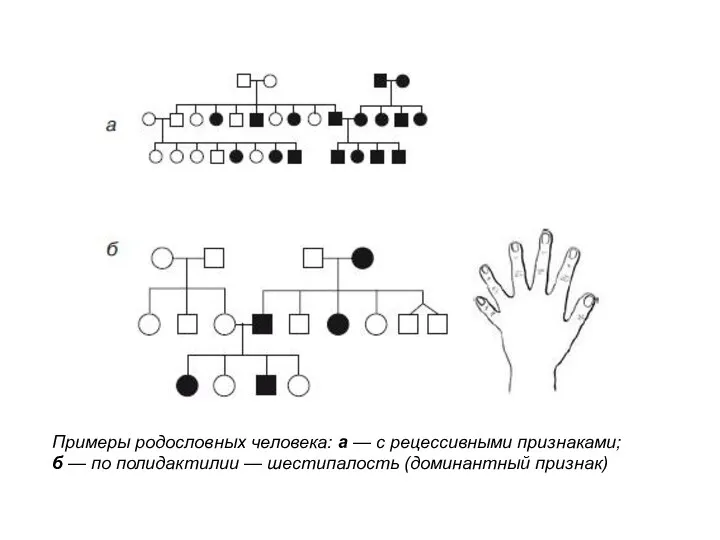
Примеры родословных человека: а — с рецессивными признаками;
б — по полидактилии — шестипалость (доминантный признак)
Примеры родословных человека: а — с рецессивными признаками;
б — по полидактилии — шестипалость (доминантный признак)
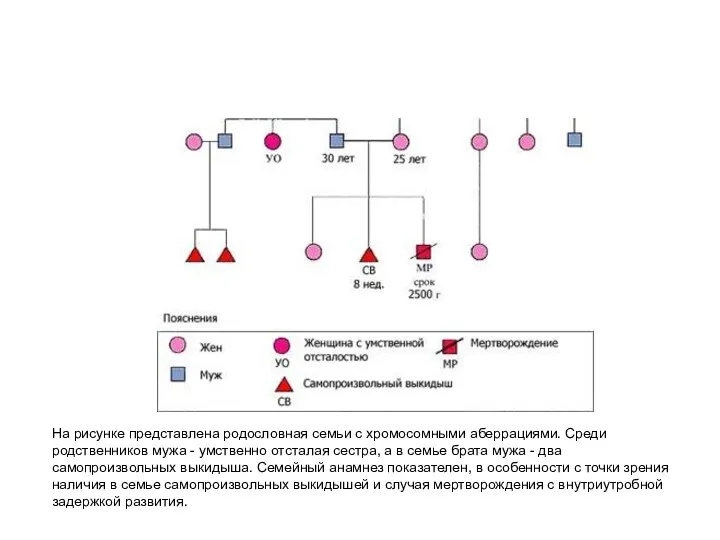
На рисунке представлена родословная семьи с хромосомными аберрациями. Среди родственников мужа
На рисунке представлена родословная семьи с хромосомными аберрациями. Среди родственников мужа
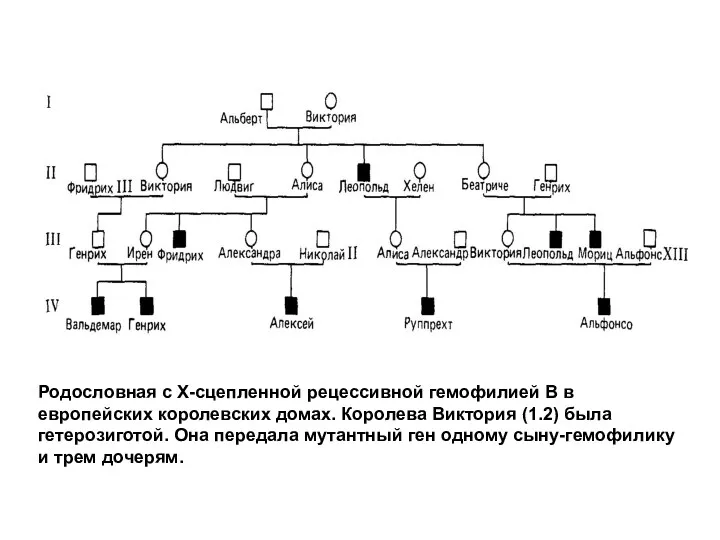
Родословная с Х-сцепленной рецессивной гемофилией В в европейских королевских домах. Королева
Родословная с Х-сцепленной рецессивной гемофилией В в европейских королевских домах. Королева

1. Сбор сведений у пробанда (лицо, к которому строится родословная) о
1. Сбор сведений у пробанда (лицо, к которому строится родословная) о

Близнецовый метод изучения генетики человека введен в медицинскую практику Ф. Гальтоном
Близнецовый метод изучения генетики человека введен в медицинскую практику Ф. Гальтоном
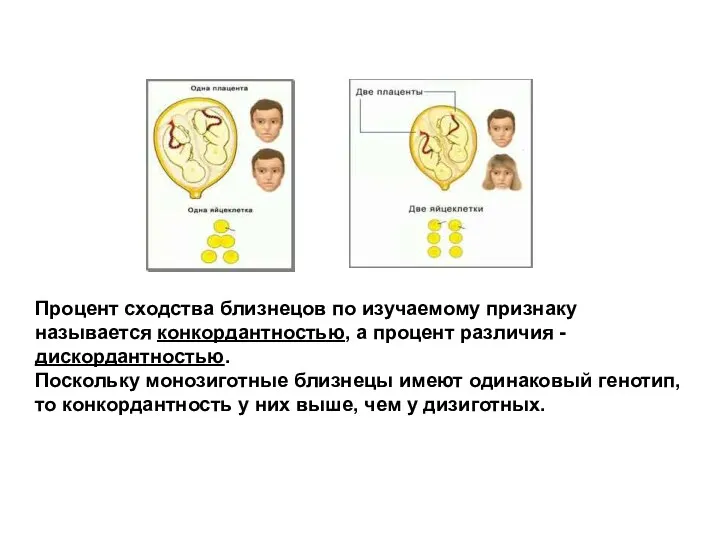
Процент сходства близнецов по изучаемому признаку называется конкордантностью, а процент различия
Процент сходства близнецов по изучаемому признаку называется конкордантностью, а процент различия

Для оценки роли наследственности и среды в развитии того или иного
Для оценки роли наследственности и среды в развитии того или иного
Пренатальная диагностика врожденных и наследственных болезней - это комплексная отрасль медицины
Пренатальная диагностика врожденных и наследственных болезней - это комплексная отрасль медицины

При организации и развитии системы пренатальной диагностики должны выполняться следующие условия:
- Диагностические процедуры должны
При организации и развитии системы пренатальной диагностики должны выполняться следующие условия: - Диагностические процедуры должны

Пренатальная диагностика должна включать два этапа:
- первый этап - выявление женщин (точнее,
Пренатальная диагностика должна включать два этапа: - первый этап - выявление женщин (точнее,

Показания к проведению пренатальной диагностики:
1. Возраст матери 35 лет;
2. Наличие в семье предыдущего
Показания к проведению пренатальной диагностики: 1. Возраст матери 35 лет; 2. Наличие в семье предыдущего
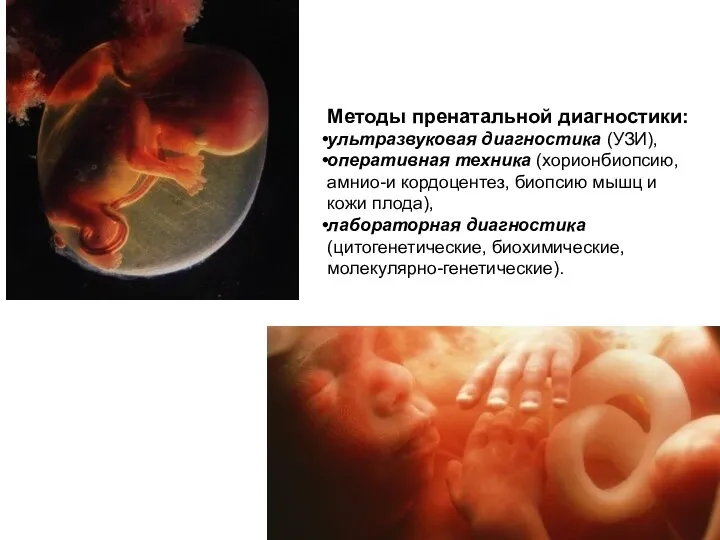
Методы пренатальной диагностики:
ультразвуковая диагностика (УЗИ),
оперативная техника (хорионбиопсию, амнио-и кордоцентез, биопсию мышц и
ультразвуковая диагностика (УЗИ),
оперативная техника (хорионбиопсию, амнио-и кордоцентез, биопсию мышц и
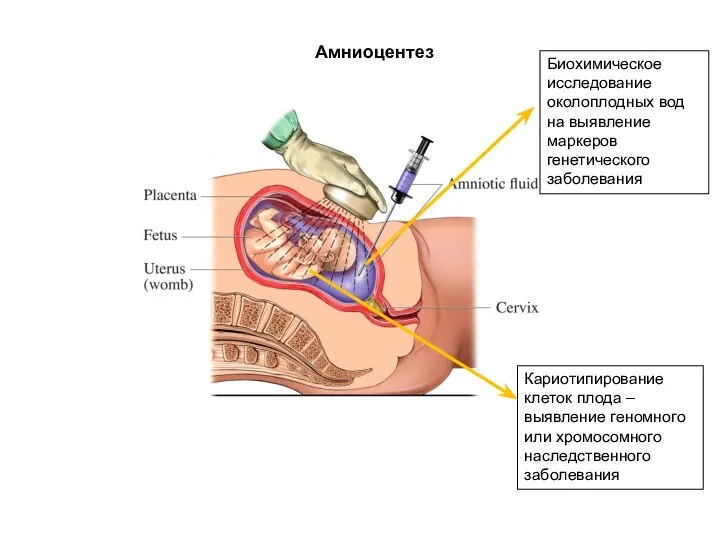
Амниоцентез
Биохимическое исследование околоплодных вод на выявление маркеров генетического заболевания
Кариотипирование клеток
Амниоцентез
Биохимическое исследование околоплодных вод на выявление маркеров генетического заболевания
Кариотипирование клеток

ЦИТОГЕНЕТИЧЕСКИЕ МЕТОДЫ
ЦИТОГЕНЕТИЧЕСКИЕ МЕТОДЫ

Цитологические методы связаны с проведением окрашивания цитологического материала и последующей микроскопией.
Цитологические методы связаны с проведением окрашивания цитологического материала и последующей микроскопией.
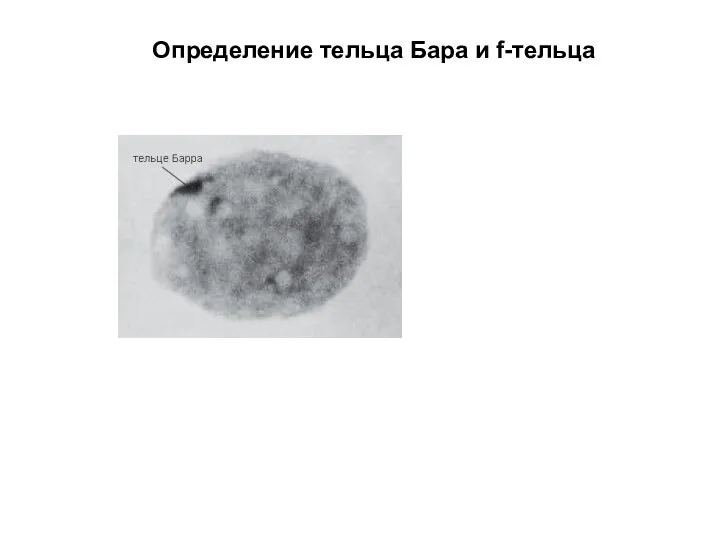
Определение тельца Бара и f-тельца
Определение тельца Бара и f-тельца
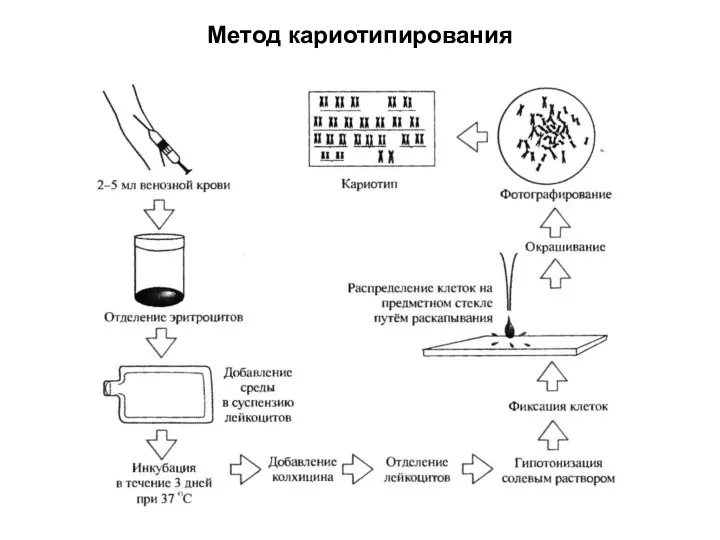
Метод кариотипирования
Метод кариотипирования

Биохимические методы. Позволяют выявить либо аномальные белки-ферменты, либо промежуточные продукты обмена, свидетельствующие
Биохимические методы. Позволяют выявить либо аномальные белки-ферменты, либо промежуточные продукты обмена, свидетельствующие
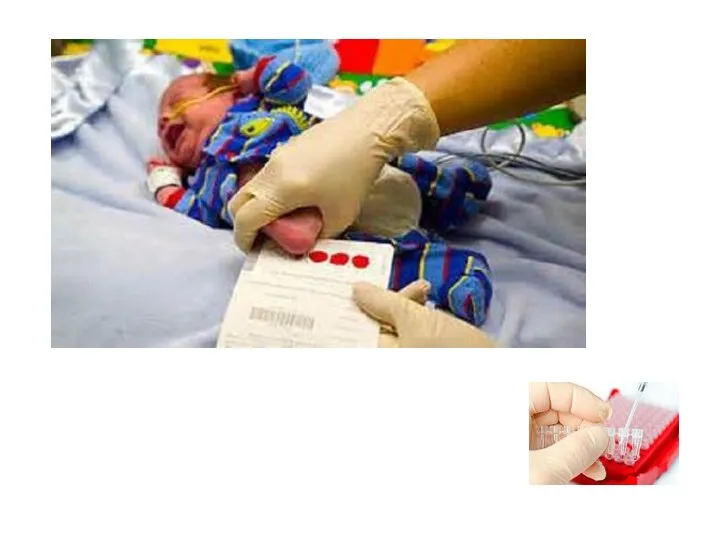

ДНК-диагностика
Это наиболее точный метод диагностики моногенных наследственных заболеваний.
Преимущества метода:
1) позволяет определить
ДНК-диагностика
Это наиболее точный метод диагностики моногенных наследственных заболеваний.
Преимущества метода:
1) позволяет определить

АКСИОМЫ МЕДЕЦИНСКОЙ ГЕНЕТИКИ
Наследственные болезни являются общей частью наследственной изменчивости человека.
В развитии
АКСИОМЫ МЕДЕЦИНСКОЙ ГЕНЕТИКИ
Наследственные болезни являются общей частью наследственной изменчивости человека.
В развитии

Ответить на вопросы:
Какие причины могут лежать в основе синдрома Дауна?
Что такое
Ответить на вопросы:
Какие причины могут лежать в основе синдрома Дауна?
Что такое
 Доброкачественные и злокачественные опухоли почек
Доброкачественные и злокачественные опухоли почек Юношеские депрессии
Юношеские депрессии Менингококки и гонококки
Менингококки и гонококки Современные возможности фитотерапии артериальной гипертензии
Современные возможности фитотерапии артериальной гипертензии Увеальная меланома
Увеальная меланома Аневризмы
Аневризмы Основные зрительные функции, особенности их развития у детей. Центральное зрение: характеристика и методы исследования
Основные зрительные функции, особенности их развития у детей. Центральное зрение: характеристика и методы исследования Equipment and instruments of dental clinic. Passive voice
Equipment and instruments of dental clinic. Passive voice Ведение пациентов с внебольничной пневмонией на амбулаторном этапе
Ведение пациентов с внебольничной пневмонией на амбулаторном этапе ОРВИ и грипп
ОРВИ и грипп Нарушения водно-солевого обмена
Нарушения водно-солевого обмена Вопросы безопасности в анестезиологии и реаниматологии. Или как анестезиологу выжить самому и сохранить жизнь пациенту
Вопросы безопасности в анестезиологии и реаниматологии. Или как анестезиологу выжить самому и сохранить жизнь пациенту Наборы инструментов для операций
Наборы инструментов для операций Вирусология. Клиническая и экологическая микробиология. Инфекция и иммунитет при вирусных заболеваниях. (Модуль 3.22)
Вирусология. Клиническая и экологическая микробиология. Инфекция и иммунитет при вирусных заболеваниях. (Модуль 3.22) Адам ағзасының қалыпты микрофлорасы. Дисбактериоз
Адам ағзасының қалыпты микрофлорасы. Дисбактериоз Грипп и его профилактика
Грипп и его профилактика Мышцы живота и спины, паховый канал
Мышцы живота и спины, паховый канал Правила личной гигиены и здоровье
Правила личной гигиены и здоровье Приобретенные (вторичные) иммунодефициты
Приобретенные (вторичные) иммунодефициты Қаңқа сүйектерінің жастық ерекшеліктерін тану
Қаңқа сүйектерінің жастық ерекшеліктерін тану Группы крови. Резус-фактор
Группы крови. Резус-фактор Статистические показатели оценки деятельности учреждений здравоохранения
Статистические показатели оценки деятельности учреждений здравоохранения Общая патофизиология
Общая патофизиология Особенности оказания сестринской помощи пациентам с хроническим бронхитом в поликлинических условиях
Особенности оказания сестринской помощи пациентам с хроническим бронхитом в поликлинических условиях Окружающая среда и здоровье человека
Окружающая среда и здоровье человека Цитомегаловирус CMV (ЦМВ)
Цитомегаловирус CMV (ЦМВ) Физиология микроциркуляции. Особенности кровообращения в различных сосудистых областях
Физиология микроциркуляции. Особенности кровообращения в различных сосудистых областях 2. Морфология нарушений обмена белков и липидо
2. Морфология нарушений обмена белков и липидо