Наследственные болезни человека. Медико-генетическое консультирование. Дородовая диагностика. Лекция 9 презентация
- Наследственные болезни человека. Медико-генетическое консультирование. Дородовая диагностика. Лекция 9

Содержание
- 2. Определение Наследственные болезни — заболевания человека, обусловленные повреждением (мутациями) наследственного аппарата (генома) клетки. . Наследственные болезни
- 3. Классификация НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ГЕННЫЕ ХРОМОСОМНЫЕ МИТОХОНД- РИАЛЬНЫЕ МОНОГЕННЫЕ ПОЛИГЕННЫЕ Наследственные болезни
- 4. Медико-генетическое консультирование - Специализированный вид медицинской помощи, направленный на предупреждение появления в семьях больных детей Проспективное
- 5. В ходе консультации семья должна получить ответы на следующие вопросы: Какова природа заболевания? (Не всякое врожденное
- 6. Задачами врача-генетика является: Поставить диагноз Рассчитать генетический риск Донести информацию до семьи (Показателем того, что консультация
- 7. Этапы МГК Сбор генетического анамнеза и построение генеалогического древа Осмотр пробанда (и его родственников) - анализ
- 8. Этап 1 1. Составление родословной Это процесс активный, у семьи выспрашивают все подробности родства, были ли
- 9. Затем, 2. Анализ фенотипа. Генетик особое внимание уделяет деталям строения и мелким анатомическим особенностям. У генетиков
- 10. ДАВИДЕНКОВ СЕРГЕЙ НИКОЛАЕВИЧ (25.08.1880 - 2.07.1961); - крупнейший невропатолог и генетик человека. В области медицинской генетики
- 11. ЭТАП 2. анализ фенотипа
- 12. Наиболее тщательно изучаются лицо, глаза, Антимонголоидный разрез глаз, гипертелоризм, телекант, гетерохромия радужек Микроцефалия, монголоидный разрез глаз
- 13. Челюсти, ротовая полость, микрогнатия макроглоссия олигодонтия Аномальные уздечки во рту «Готическое» нёбо
- 14. Уши, микротия Периаурикулярные выросты Атрезия слухового прохода Периарикулярные ямки и складки Низко посаженные уши
- 15. Кисти и стопы
- 16. Клинодактилия мизинца арахнодактилия камптодактилия Брахидактилия и клинодактилия Брахи- и синдактилия
- 17. Кожа, ногти, волосы Гемангиома лица при синдроме Штурге-Вебера витилиго Сверхрастяжимость и рубцы типа «папиросной бумаги» при
- 18. Анализ фенотипа позволяет предположить диагноз Но иногда требуются дополнительные исследования: Кариотипирование Поиск подобного фенотипа в литературе
- 19. Второе посещение Постановка диагноза Расчет риска Донесение информации до семьи Наблюдение и лечение В случае беременности
- 20. Итак, диагноз. Каковы же основные группы наследственных заболеваний? Моногенные, или менделирующие болезни , когда заболевание определяется
- 21. Генные болезни Генные болезни - это группа заболеваний, обусловленных мутациями на генном уровне. Общая частота генных
- 22. Генные Изменения одного гена. Нуклеотиды, составляющие ген, могут «выпадать», меняться местами, заменять А на Т. Причинами
- 23. Моногенные болезни Наследственные болезни Вызваны мутациями или отсутствием отдельных генов. Наследуются в полном соответствии с законами
- 24. 1. Моногенные болезни разнообразны: Ферментопатии – дефекты отдельных ферментов Дисплазии – нарушение строения тканей Синдромы МВПР
- 25. Синдром Марфана Наследственная болезнь соединительной ткани, вызванная мутацией гена, кодирующего структуру белка фибриллина. Наследуется по аутосомно-доминантному
- 26. Известные люди с синдромом Марфана Эхнатон Н. Паганини Ш. де Голль А. Линкольн Наследственные болезни
- 27. Муковисцидоз Заболевание, при котором поражаются экзокринные железы. Причина - мутация (делеция трех нуклеотидов), приводящая к отсутствию
- 28. Ферментопатии (синдромы дизметаболизма) – дефект отдельного фермента, например: Фенилкетонурия, АР Адреногенитальный синдром, АР Мукополисахаридозы Дети рождаются
- 29. Дисплазии (при мутациях генов, экспрессирующихся в определенных тканях): Нейрофиброматоз (болезнь Реклингаузена), АД Ахондроплазия, АД Синдром Марфана,
- 30. Признаки нейрофиброматоза: фибромы и пятна типа «кофе с молоком». Фибромы происходят из Шванновских клеток. И меланоциты
- 31. Синдромы МВПР (множественных врожденных пороков развития) как результат мутаций важных регуляторных или генов с плейотропным эффектом
- 32. Синдром Алажилля – аутосомно-доминантный синдром Характерное лицо Внутрипеченочный холестаз Врожденный порок сердца Дефекты глазного яблока Малые
- 33. Генетический риск при моногенных болезнях, передающихся в семье, определяется по законам Менделя Аа Аа АА Аа
- 34. Если заболевание регистрируется в семье впервые – это свидетельствует о новой мутации. Риск для следующего ребенка
- 35. Частота встречаемости разных моногенных болезней в европейской популяции АД Семейная гиперхолестеринемия 1 : 500 Поликистоз почек
- 36. Доля новых мутаций для некоторых моногенных болезней: Ахондроплазия – 80% Нейрофиброматоз – 40% Синдром Марфана –
- 37. 2. Хромосомные болезни Группа болезней, в основе развития которых лежат нарушения числа или структуры хромосом, возникающие
- 38. Хромосомные мутации Затрагивают участки хромосом или целые хромосомы, меняют структуру, форму. Происходят при кроссинговере – перекрёсте
- 39. Геномные мутации Связаны с изменением числа хромосом внутри генома. Часто происходят при ошибочном выстраивании веретена деления
- 40. Генетический риск при хромосомных болезнях рассчитывается исходя из цитогенетической картины Появление хромосомной или геномной мутации у
- 41. Причины болезней связанные с нарушением плоидности вызванные нарушением числа хромосом связанные с изменением структуры хромосом. ХРОМОСОМНЫЕ
- 42. Нарушение плоидности Наследственные болезни Геномные мутации - изменения количества хромосом в геноме Анеуплоидии – изменение числа
- 43. Например, классический вариант синдрома Дауна (95% случаев) – полная трисомия (то есть трисомия во всех клетках)
- 44. 2. Семейные транслокации дают высокий риск Пара 14 Пара 21 Родитель со сбалансированной транслокацией 14\21 гамета
- 45. При слиянии гамет только один потомок получит нормальный набор Не совместимы с жизнью Транслокаци-онный синдром Дауна
- 46. Бывает риск даже 100% Например, при транслокации 21-й хромосомы на ее гомолог, риск рождения больного ребенка
- 47. Итак, при определении риска при хромосомной патологии у ребенка следует исходить из кариотипа родителей
- 48. Формы анеуплоидий Моносомия — наличие в генотипе всего одной из пары гомологичных хромосом. Моносомия по половой
- 49. Формы анеуплоидий Трисомия - наличие в клетке одной дополнительной хромосомы вместо обычного (диплоидного) хромосомного набора. Известные
- 50. Синдром Эдвардса Наследственные болезни Кариотип человека с синдром трисомии 18
- 51. Мир равных возможностей Синдром Дауна – не трагедия, если тебя любят! 21 марта – Международный день
- 52. Трисомии по половым хромосомам Синдром Клайнфельтера - трисомия по Х хромосоме (47,XXY, ХХХУ, ХУУ и т.д.).
- 53. Изменения структуры хромосом Рис. 1. Транслокации между Рис. 2. Делеция 8-й и 11-й хромосомами части длинного
- 54. Болезни хромосомных перестроек Транслокация 46 ХХ, t(4;13)(q25; q22) приводит к задержке психоречевого развития, множественным порокам развития;
- 55. 3. Митохондриальные болезни Затрагивают гены митохондрий. Известно около 30 болезней. Синдром Лебера (1988) - проявляется быстрым
- 56. Наследование мт ДНК Наследственные болезни
- 57. 4. Мультифакториальные заболевания Обусловлены как наследственными факторами, так и факторами внешней среды. Это наиболее распространенные болезни:
- 58. Полигенные болезни Обусловлены взаимодействием определенных комбинаций аллелей разных локусов и внешних факторов. Не наследуются по законам
- 59. Схема мультифакториального заболевания ген ген ген ген ген ген ген ген ген среда Мультифакториальная болезнь
- 60. Пример: Упрощенная схема развития бронхиальной астмы (Бронхогенной гиперреактивности)
- 61. Не для запоминания!
- 63. 1900, а?
- 64. Факторы среды, провоцирующие бронхиальную астму пыльца плесень домашние животные пылевые клещи
- 65. Обобщенные данные литературы по мультифакториальным заболеваниям собраны в так называемые таблицам эмпирического риска Риск в таких
- 66. Другие гены регуляторы гомеостаза Риск заболевания родственников больных с различными психическими болезнями (в процентах) Пример такой
- 67. Итак, врач тем или иным способом рассчитывает риск появления заболевания у потомства Риск развития заболевания менее
- 68. Заключительный третий этап консультирования – сообщение результатов семье Сообщается только родителям На беседу отводится столько времени,
- 69. Решением родителей может стать: Рожать Не рожать Усыновить Разорвать брак Родить от другого партнера Применить донорское
- 70. Только взглянуть!
- 71. Дородовая (пренатальная) диагностика
- 72. методы дородовой диагностики Неинвазивные методы: Ультразвуковое исследование (во все сроки) ХГЧ, альфа-фетопротеин и эстриол в крови
- 73. Предимплантационная диагностика При экстракорпоральном оплодотворении (ЭКО) берутся бластомеры на стадии морулы и изучаются до имплантации зародыша
- 74. Неинвазивные методы УЗИ Исследование сыворотки матери
- 75. Инвазивные методы
- 76. Биопсия хориона на 8 – 10 неделе беременности
- 77. Кордоцентез – взятие крови из пупочной вены Амниоцентез – взятие околоплодных вод Плацентоцентез – биопсия ткани
- 78. Процедуры проводят под контролем УЗИ
- 79. Возможностей немало
- 82. Скачать презентацию

Определение
Наследственные болезни — заболевания человека, обусловленные повреждением (мутациями) наследственного аппарата (генома)
Определение
Наследственные болезни — заболевания человека, обусловленные повреждением (мутациями) наследственного аппарата (генома)
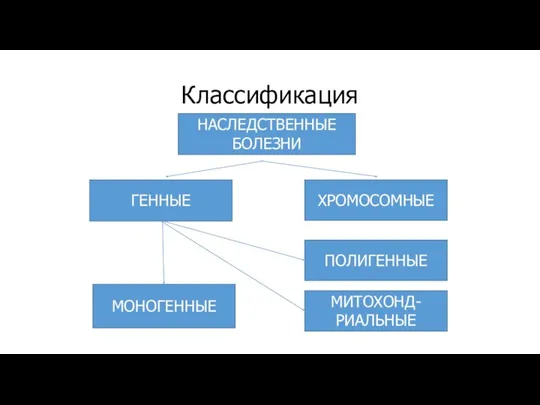
Классификация
НАСЛЕДСТВЕННЫЕ
БОЛЕЗНИ
ГЕННЫЕ
ХРОМОСОМНЫЕ
МИТОХОНД-
РИАЛЬНЫЕ
МОНОГЕННЫЕ
ПОЛИГЕННЫЕ
Наследственные болезни
Классификация
НАСЛЕДСТВЕННЫЕ
БОЛЕЗНИ
ГЕННЫЕ
ХРОМОСОМНЫЕ
МИТОХОНД-
РИАЛЬНЫЕ
МОНОГЕННЫЕ
ПОЛИГЕННЫЕ
Наследственные болезни

Медико-генетическое консультирование -
Специализированный вид медицинской помощи, направленный на предупреждение появления
Медико-генетическое консультирование -
Специализированный вид медицинской помощи, направленный на предупреждение появления

В ходе консультации семья должна получить ответы на следующие вопросы:
Какова природа
В ходе консультации семья должна получить ответы на следующие вопросы:
Какова природа

Задачами врача-генетика является:
Поставить диагноз
Рассчитать генетический риск
Донести информацию до семьи
(Показателем
Задачами врача-генетика является:
Поставить диагноз
Рассчитать генетический риск
Донести информацию до семьи
(Показателем

Этапы МГК
Сбор генетического анамнеза и построение генеалогического древа
Осмотр пробанда (и его
Этапы МГК
Сбор генетического анамнеза и построение генеалогического древа
Осмотр пробанда (и его

Этап 1
1. Составление родословной
Это процесс активный, у семьи выспрашивают все подробности
Этап 1
1. Составление родословной
Это процесс активный, у семьи выспрашивают все подробности

Затем,
2. Анализ фенотипа.
Генетик особое внимание уделяет деталям строения и мелким анатомическим
Затем,
2. Анализ фенотипа.
Генетик особое внимание уделяет деталям строения и мелким анатомическим

ДАВИДЕНКОВ СЕРГЕЙ НИКОЛАЕВИЧ (25.08.1880 - 2.07.1961);
- крупнейший невропатолог и генетик
ДАВИДЕНКОВ СЕРГЕЙ НИКОЛАЕВИЧ (25.08.1880 - 2.07.1961);
- крупнейший невропатолог и генетик

ЭТАП 2. анализ фенотипа
ЭТАП 2. анализ фенотипа
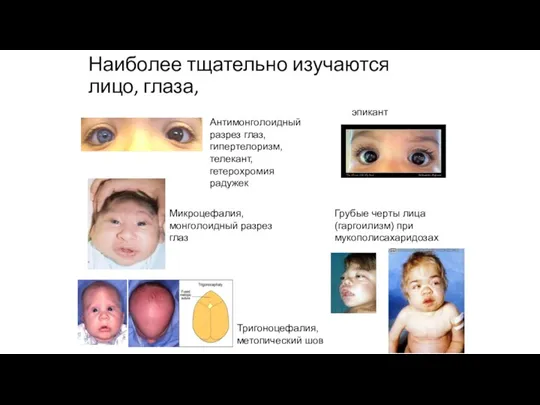
Наиболее тщательно изучаются лицо, глаза,
Антимонголоидный разрез глаз, гипертелоризм, телекант, гетерохромия радужек
Микроцефалия,
Наиболее тщательно изучаются лицо, глаза,
Антимонголоидный разрез глаз, гипертелоризм, телекант, гетерохромия радужек
Микроцефалия,
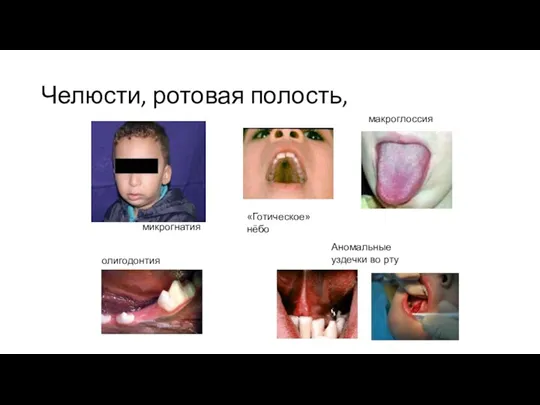
Челюсти, ротовая полость,
микрогнатия
макроглоссия
олигодонтия
Аномальные уздечки во рту
«Готическое» нёбо
Челюсти, ротовая полость,
микрогнатия
макроглоссия
олигодонтия
Аномальные уздечки во рту
«Готическое» нёбо
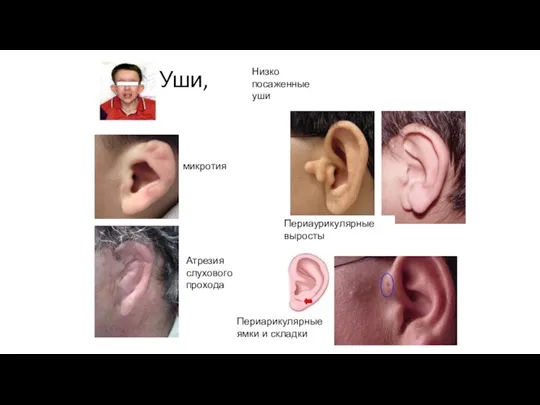
Уши,
микротия
Периаурикулярные выросты
Атрезия слухового прохода
Периарикулярные ямки и складки
Низко посаженные уши
Уши,
микротия
Периаурикулярные выросты
Атрезия слухового прохода
Периарикулярные ямки и складки
Низко посаженные уши
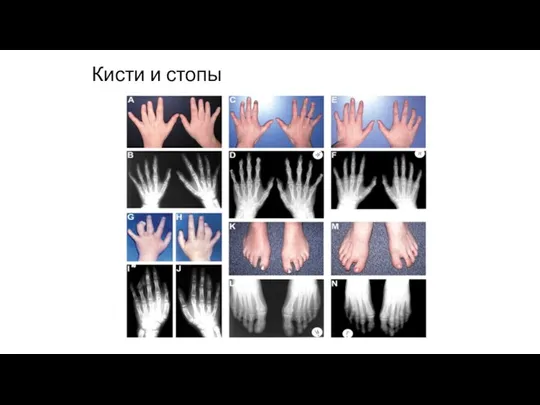
Кисти и стопы
Кисти и стопы
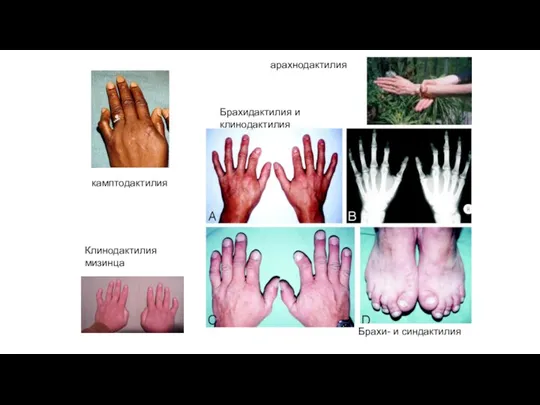
Клинодактилия мизинца
арахнодактилия
камптодактилия
Брахидактилия и клинодактилия
Брахи- и синдактилия
Клинодактилия мизинца
арахнодактилия
камптодактилия
Брахидактилия и клинодактилия
Брахи- и синдактилия
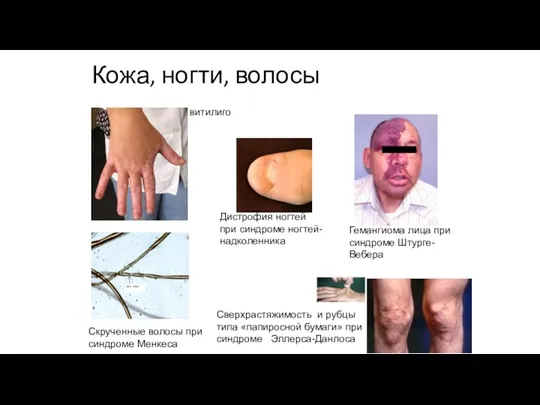
Кожа, ногти, волосы
Гемангиома лица при синдроме Штурге-Вебера
витилиго
Сверхрастяжимость и рубцы типа «папиросной
Кожа, ногти, волосы
Гемангиома лица при синдроме Штурге-Вебера
витилиго
Сверхрастяжимость и рубцы типа «папиросной

Анализ фенотипа позволяет предположить диагноз
Но иногда требуются дополнительные исследования:
Кариотипирование
Поиск подобного фенотипа
Анализ фенотипа позволяет предположить диагноз
Но иногда требуются дополнительные исследования:
Кариотипирование
Поиск подобного фенотипа

Второе посещение
Постановка диагноза
Расчет риска
Донесение информации до семьи
Наблюдение и лечение
В случае беременности
Второе посещение
Постановка диагноза
Расчет риска
Донесение информации до семьи
Наблюдение и лечение
В случае беременности

Итак, диагноз. Каковы же основные группы наследственных заболеваний?
Моногенные, или менделирующие болезни
Итак, диагноз. Каковы же основные группы наследственных заболеваний?
Моногенные, или менделирующие болезни

Генные болезни
Генные болезни - это группа заболеваний, обусловленных мутациями на
Генные болезни
Генные болезни - это группа заболеваний, обусловленных мутациями на

Генные
Изменения одного гена. Нуклеотиды, составляющие ген, могут «выпадать», меняться местами, заменять
Генные
Изменения одного гена. Нуклеотиды, составляющие ген, могут «выпадать», меняться местами, заменять

Моногенные болезни
Наследственные болезни
Вызваны мутациями или
отсутствием отдельных генов.
Моногенные болезни
Наследственные болезни
Вызваны мутациями или
отсутствием отдельных генов.

1. Моногенные болезни разнообразны:
Ферментопатии – дефекты отдельных ферментов
Дисплазии – нарушение строения
1. Моногенные болезни разнообразны:
Ферментопатии – дефекты отдельных ферментов
Дисплазии – нарушение строения
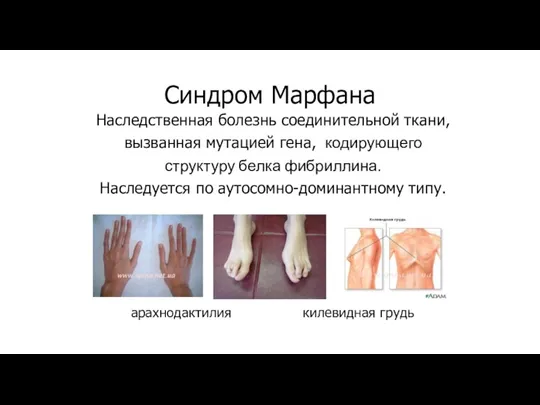
Синдром Марфана
Наследственная болезнь соединительной ткани,
вызванная мутацией гена, кодирующего
структуру белка фибриллина.
Наследуется по
Синдром Марфана
Наследственная болезнь соединительной ткани,
вызванная мутацией гена, кодирующего
структуру белка фибриллина.
Наследуется по

Известные люди с синдромом Марфана
Эхнатон Н. Паганини
Ш. де Голль А.
Известные люди с синдромом Марфана
Эхнатон Н. Паганини
Ш. де Голль А.
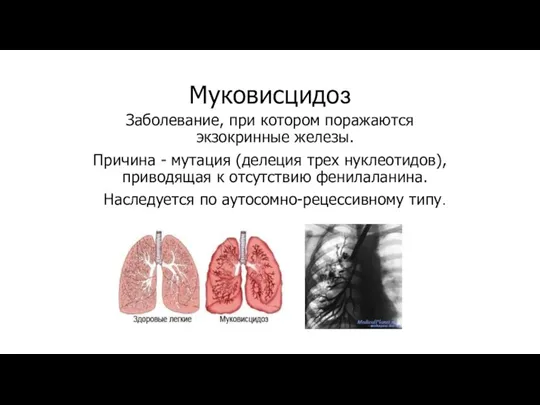
Муковисцидоз
Заболевание, при котором поражаются экзокринные железы.
Причина - мутация (делеция трех
Муковисцидоз
Заболевание, при котором поражаются экзокринные железы.
Причина - мутация (делеция трех

Ферментопатии
(синдромы дизметаболизма) – дефект отдельного фермента, например:
Фенилкетонурия, АР
Адреногенитальный синдром, АР
Мукополисахаридозы
Ферментопатии
(синдромы дизметаболизма) – дефект отдельного фермента, например:
Фенилкетонурия, АР
Адреногенитальный синдром, АР
Мукополисахаридозы

Дисплазии
(при мутациях генов, экспрессирующихся в определенных тканях):
Нейрофиброматоз (болезнь Реклингаузена), АД
Ахондроплазия,
Дисплазии
(при мутациях генов, экспрессирующихся в определенных тканях):
Нейрофиброматоз (болезнь Реклингаузена), АД
Ахондроплазия,
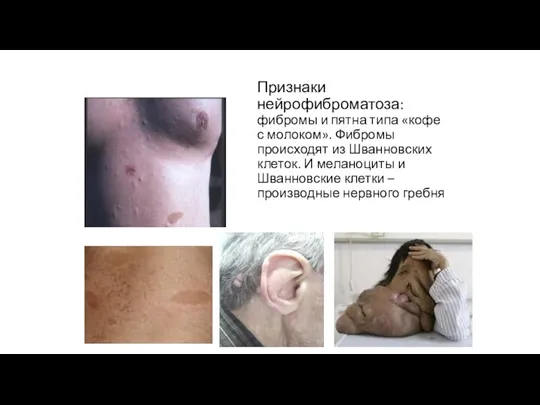
Признаки нейрофиброматоза:
фибромы и пятна типа «кофе с молоком». Фибромы происходят
Признаки нейрофиброматоза: фибромы и пятна типа «кофе с молоком». Фибромы происходят

Синдромы МВПР
(множественных врожденных пороков развития) как результат мутаций важных регуляторных
Синдромы МВПР (множественных врожденных пороков развития) как результат мутаций важных регуляторных

Синдром Алажилля – аутосомно-доминантный синдром
Характерное лицо
Внутрипеченочный холестаз
Врожденный порок
Синдром Алажилля – аутосомно-доминантный синдром
Характерное лицо
Внутрипеченочный холестаз
Врожденный порок

Генетический риск при моногенных болезнях, передающихся в семье, определяется по законам
Генетический риск при моногенных болезнях, передающихся в семье, определяется по законам

Если заболевание регистрируется в семье впервые – это свидетельствует о новой
Если заболевание регистрируется в семье впервые – это свидетельствует о новой

Частота встречаемости разных моногенных болезней в европейской популяции
АД
Семейная гиперхолестеринемия 1 : 500
Поликистоз
Частота встречаемости разных моногенных болезней в европейской популяции
АД
Семейная гиперхолестеринемия 1 : 500
Поликистоз

Доля новых мутаций для некоторых моногенных болезней:
Ахондроплазия – 80%
Нейрофиброматоз – 40%
Синдром
Доля новых мутаций для некоторых моногенных болезней:
Ахондроплазия – 80%
Нейрофиброматоз – 40%
Синдром

2. Хромосомные болезни
Группа болезней, в основе развития которых лежат нарушения
2. Хромосомные болезни
Группа болезней, в основе развития которых лежат нарушения

Хромосомные мутации
Затрагивают участки хромосом или целые хромосомы, меняют структуру, форму.
Хромосомные мутации
Затрагивают участки хромосом или целые хромосомы, меняют структуру, форму.

Геномные мутации
Связаны с изменением числа хромосом внутри генома. Часто происходят при
Геномные мутации
Связаны с изменением числа хромосом внутри генома. Часто происходят при

Генетический риск при хромосомных болезнях рассчитывается исходя из цитогенетической картины
Появление хромосомной
Генетический риск при хромосомных болезнях рассчитывается исходя из цитогенетической картины
Появление хромосомной
Причины болезней
связанные с нарушением
плоидности
вызванные нарушением
числа хромосом
связанные с
Причины болезней
связанные с нарушением
плоидности
вызванные нарушением
числа хромосом
связанные с
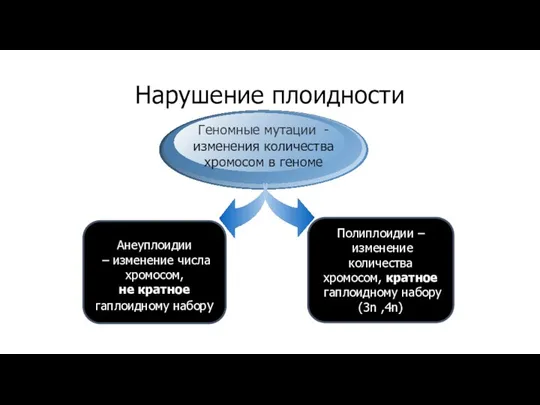
Нарушение плоидности
Наследственные болезни
Геномные мутации - изменения количества
хромосом в геноме
Анеуплоидии
Нарушение плоидности
Наследственные болезни
Геномные мутации - изменения количества
хромосом в геноме
Анеуплоидии

Например, классический вариант синдрома Дауна (95% случаев) – полная трисомия (то
Например, классический вариант синдрома Дауна (95% случаев) – полная трисомия (то
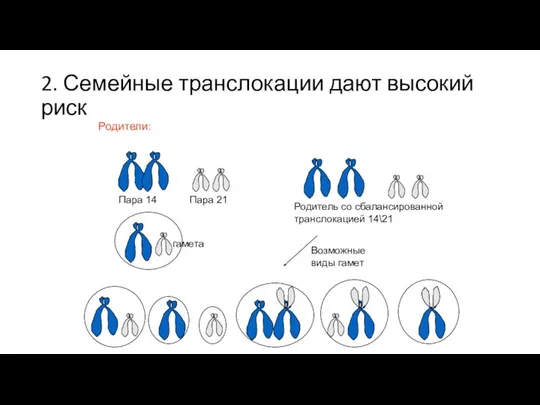
2. Семейные транслокации дают высокий риск
Пара 14
Пара 21
Родитель со сбалансированной транслокацией
2. Семейные транслокации дают высокий риск
Пара 14
Пара 21
Родитель со сбалансированной транслокацией
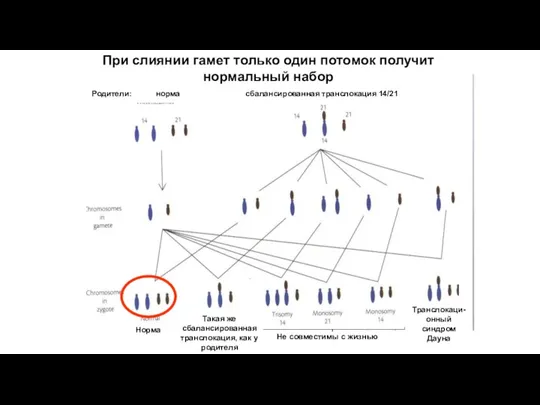
При слиянии гамет только один потомок получит нормальный набор
Не совместимы
При слиянии гамет только один потомок получит нормальный набор
Не совместимы

Бывает риск даже 100%
Например, при транслокации 21-й хромосомы на ее гомолог,
Бывает риск даже 100%
Например, при транслокации 21-й хромосомы на ее гомолог,

Итак, при определении риска при хромосомной патологии у ребенка следует исходить
Итак, при определении риска при хромосомной патологии у ребенка следует исходить

Формы анеуплоидий
Моносомия — наличие в генотипе всего одной из пары гомологичных
Формы анеуплоидий
Моносомия — наличие в генотипе всего одной из пары гомологичных

Формы анеуплоидий
Трисомия - наличие в клетке одной дополнительной хромосомы вместо обычного
Формы анеуплоидий
Трисомия - наличие в клетке одной дополнительной хромосомы вместо обычного

Синдром Эдвардса
Наследственные болезни
Кариотип человека с синдром трисомии 18
Синдром Эдвардса
Наследственные болезни
Кариотип человека с синдром трисомии 18

Мир равных возможностей
Синдром Дауна – не трагедия, если тебя любят!
21 марта
Мир равных возможностей
Синдром Дауна – не трагедия, если тебя любят!
21 марта

Трисомии по половым хромосомам
Синдром Клайнфельтера - трисомия по
Х хромосоме
Трисомии по половым хромосомам
Синдром Клайнфельтера - трисомия по
Х хромосоме

Изменения структуры хромосом
Рис. 1. Транслокации между Рис. 2. Делеция
8-й и
Изменения структуры хромосом
Рис. 1. Транслокации между Рис. 2. Делеция
8-й и

Болезни хромосомных перестроек
Транслокация 46 ХХ, t(4;13)(q25; q22) приводит к
Болезни хромосомных перестроек
Транслокация 46 ХХ, t(4;13)(q25; q22) приводит к

3. Митохондриальные болезни
Затрагивают гены митохондрий.
Известно около 30 болезней.
Синдром Лебера (1988) -
3. Митохондриальные болезни
Затрагивают гены митохондрий.
Известно около 30 болезней.
Синдром Лебера (1988) -

Наследование мт ДНК
Наследственные болезни
Наследование мт ДНК
Наследственные болезни

4. Мультифакториальные заболевания
Обусловлены как наследственными факторами, так и факторами внешней среды.
4. Мультифакториальные заболевания
Обусловлены как наследственными факторами, так и факторами внешней среды.

Полигенные болезни
Обусловлены взаимодействием определенных комбинаций аллелей разных локусов и внешних
Полигенные болезни
Обусловлены взаимодействием определенных комбинаций аллелей разных локусов и внешних
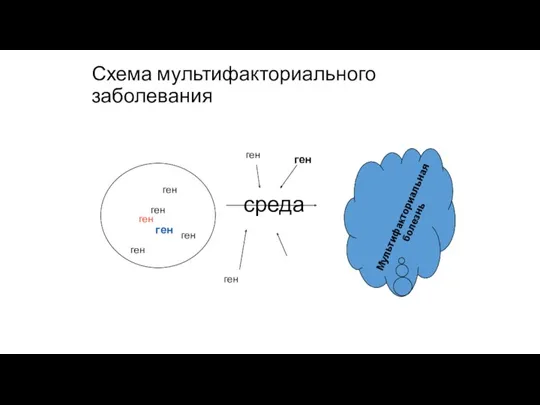
Схема мультифакториального заболевания
ген
ген
ген
ген
ген
ген
ген
ген
ген
среда
Мультифакториальная
болезнь
Схема мультифакториального заболевания
ген
ген
ген
ген
ген
ген
ген
ген
ген
среда
Мультифакториальная
болезнь
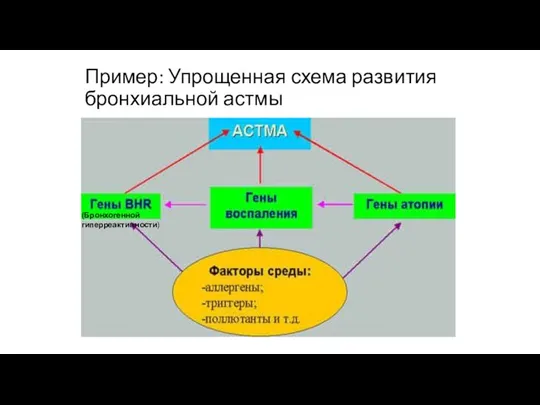
Пример: Упрощенная схема развития бронхиальной астмы
(Бронхогенной гиперреактивности)
Пример: Упрощенная схема развития бронхиальной астмы
(Бронхогенной гиперреактивности)
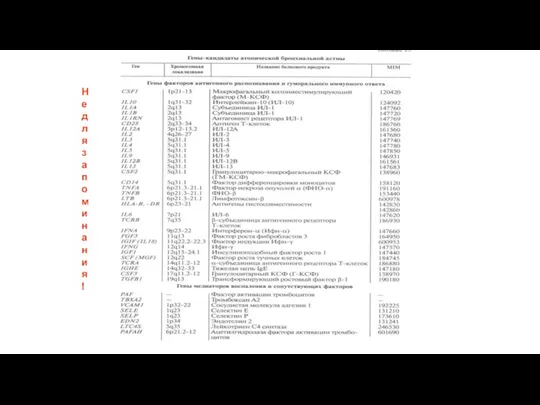
Не для запоминания!
Не для запоминания!


1900, а?
1900, а?
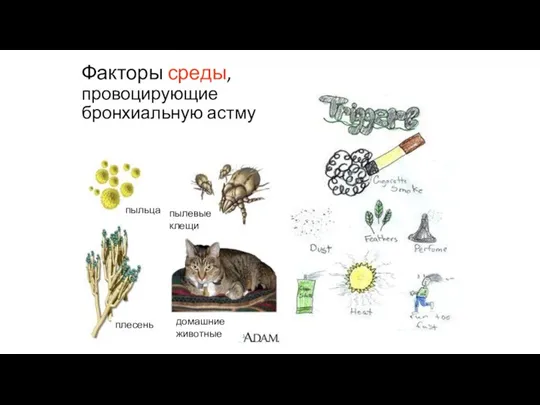
Факторы среды, провоцирующие бронхиальную астму
пыльца
плесень
домашние животные
пылевые клещи
Факторы среды, провоцирующие бронхиальную астму
пыльца
плесень
домашние животные
пылевые клещи

Обобщенные данные литературы по мультифакториальным заболеваниям собраны в так называемые таблицам
Обобщенные данные литературы по мультифакториальным заболеваниям собраны в так называемые таблицам
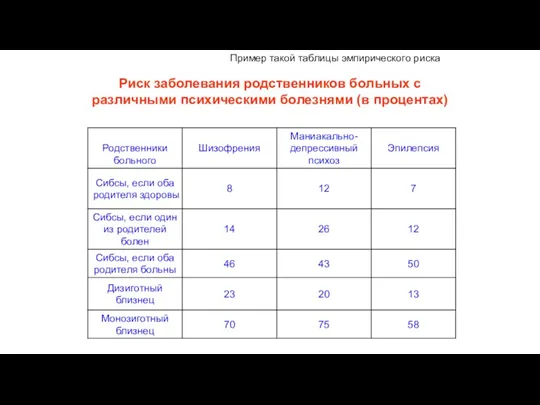
Другие гены регуляторы гомеостаза
Риск заболевания родственников больных с различными психическими болезнями
Другие гены регуляторы гомеостаза
Риск заболевания родственников больных с различными психическими болезнями

Итак, врач тем или иным способом рассчитывает риск появления заболевания у
Итак, врач тем или иным способом рассчитывает риск появления заболевания у

Заключительный третий этап консультирования – сообщение результатов семье
Сообщается только родителям
На беседу
Заключительный третий этап консультирования – сообщение результатов семье
Сообщается только родителям
На беседу

Решением родителей может стать:
Рожать
Не рожать
Усыновить
Разорвать брак
Родить от другого партнера
Применить донорское
Решением родителей может стать:
Рожать
Не рожать
Усыновить
Разорвать брак
Родить от другого партнера
Применить донорское
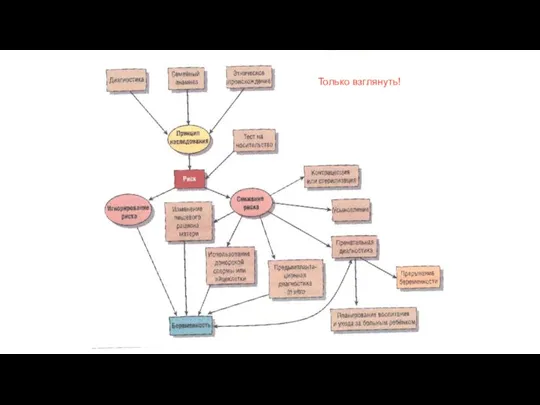
Только взглянуть!
Только взглянуть!

Дородовая (пренатальная) диагностика
Дородовая (пренатальная) диагностика

методы дородовой диагностики
Неинвазивные методы:
Ультразвуковое исследование (во все сроки)
ХГЧ, альфа-фетопротеин и
методы дородовой диагностики
Неинвазивные методы:
Ультразвуковое исследование (во все сроки)
ХГЧ, альфа-фетопротеин и

Предимплантационная диагностика
При экстракорпоральном оплодотворении (ЭКО) берутся бластомеры на стадии морулы и
Предимплантационная диагностика
При экстракорпоральном оплодотворении (ЭКО) берутся бластомеры на стадии морулы и

Неинвазивные методы
УЗИ
Исследование сыворотки матери
Неинвазивные методы
УЗИ
Исследование сыворотки матери

Инвазивные методы
Инвазивные методы

Биопсия хориона на 8 – 10 неделе беременности
Биопсия хориона на 8 – 10 неделе беременности
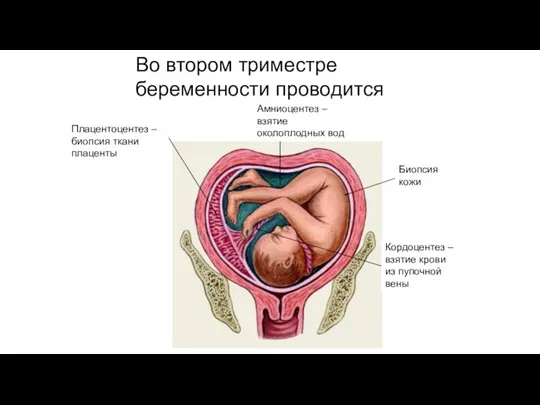
Кордоцентез – взятие крови из пупочной вены
Амниоцентез – взятие околоплодных вод
Плацентоцентез
Кордоцентез – взятие крови из пупочной вены
Амниоцентез – взятие околоплодных вод
Плацентоцентез
Процедуры проводят под контролем УЗИ
Процедуры проводят под контролем УЗИ
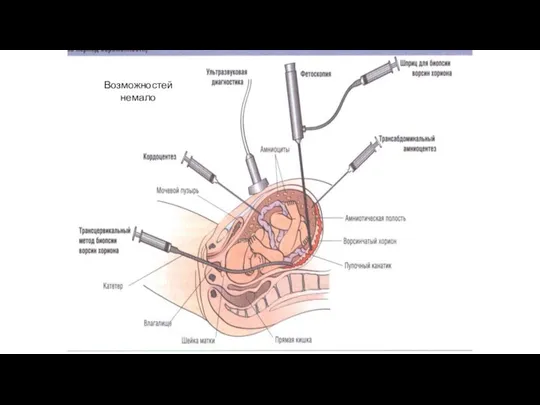
Возможностей немало
Возможностей немало
 Laryngeal edema and stenosis
Laryngeal edema and stenosis Диета терапия при заболеваниях легких
Диета терапия при заболеваниях легких Статистические показатели оценки деятельности учреждений здравоохранения
Статистические показатели оценки деятельности учреждений здравоохранения ДНҚ репарациясы
ДНҚ репарациясы Противоэпилептические и противопаркинсонические средства
Противоэпилептические и противопаркинсонические средства Профілактика бактеріальних захворювань людини
Профілактика бактеріальних захворювань людини Патологическая мышечная утомляемость
Патологическая мышечная утомляемость Розацеа. Этиология и патогенез
Розацеа. Этиология и патогенез Биполярное расстройство I типа
Биполярное расстройство I типа Симптомы поражения различных долей мозга
Симптомы поражения различных долей мозга Рак молочной железы
Рак молочной железы Анальгетические средства
Анальгетические средства БМСК ұйымдарында әріптестермен, арнайы орта медициналық қызметкерлермен
БМСК ұйымдарында әріптестермен, арнайы орта медициналық қызметкерлермен Диагностическое значение лабораторно-инструментальных методов исследований при заболеваниях суставов
Диагностическое значение лабораторно-инструментальных методов исследований при заболеваниях суставов Қан тобын анықтау. Резус фактор анықтау,қан тобының сәйкестігін анықтау. Қан құю техникасы
Қан тобын анықтау. Резус фактор анықтау,қан тобының сәйкестігін анықтау. Қан құю техникасы Красный плоский лишай (КПЛ). Псориаз (П)
Красный плоский лишай (КПЛ). Псориаз (П) Здоровье детей – простое и надёжное решение от Мастер Доктор
Здоровье детей – простое и надёжное решение от Мастер Доктор Завдання, пов'язані з медициною катастроф і військовою медициною
Завдання, пов'язані з медициною катастроф і військовою медициною Инсульт. Острое нарушение мозгового кровообращения
Инсульт. Острое нарушение мозгового кровообращения Общее о воспалении. Продуктивное и специфическое воспаление
Общее о воспалении. Продуктивное и специфическое воспаление Ерекше тұқым қуалайтын моногенді аурулар. Анықтамасы, себептері, жіктелуі, клиникалық белгілері, тұқым қуалау типтері
Ерекше тұқым қуалайтын моногенді аурулар. Анықтамасы, себептері, жіктелуі, клиникалық белгілері, тұқым қуалау типтері Патофизиология водно-электролитного обмена. Отеки
Патофизиология водно-электролитного обмена. Отеки Клиническая классификация ВИЧ-инфекции
Клиническая классификация ВИЧ-инфекции Гигиенические требования к земельному участку, зданию ДОУ и планировке помещений
Гигиенические требования к земельному участку, зданию ДОУ и планировке помещений Стимуляция овуляции в протоколах ЭКО. Осложнения при лечении бесплодия с использованием ВРТ
Стимуляция овуляции в протоколах ЭКО. Осложнения при лечении бесплодия с использованием ВРТ Поражение органа зрения при неврологических заболеваниях
Поражение органа зрения при неврологических заболеваниях Организация и принципы работы детской поликлиники
Организация и принципы работы детской поликлиники Дыхательная недостаточность. Определение
Дыхательная недостаточность. Определение