- Наследственные нейромышечные заболевания

Содержание
- 2. Понимание ведущей роли генетических факторов в этиологии и патогенезе многих болезней нервной системы сформировалось в первой
- 3. Конец XX века и начало нового тысячелетия ознаменовался бурным развитием молекулярной генетики человека, важнейшие достижения которой
- 4. В 1990-2000 годы, получившие название «Десятилетие мозга», фактически произошла смена фундаментальных концепций, касающихся молекулярных основ этиологии
- 5. Идентификация мутантных генов и первичных биохимических нарушений, ответственных за развитие многих десятков подобных заболеваний, привела к
- 6. На этой основе были сформированы новые неизвестные ранее классы заболеваний, такие как болезни экспансии, конформационные болезни
- 7. В основе болезней экспансии, лежат динамические мутации, обусловленные нестабильностью расположенных в значимых областях генов тринуклеотидных сателлитных
- 8. Патогенетический механизм нейродегенеративных болезней, обусловленных экспансией CAG-или GCG-повторов, расположенных в кодирующих областях генов, связан с изменением
- 9. Подобные болезни получили название конформационных. К ним относятся некоторые миопатии, синдромы паркинсонизма, амилоидные нейропатии, наследственные формы
- 10. При наследственных заболеваниях с пароксизмами, гипер- или гипо-возбудимостью вовлеченных в патологический процесс тканей, обнаружены мутации в
- 11. К каналопатиям относятся миотонии, периодические параличи, наследственные формы абсанс эпилепсии и мигрени
- 12. Нервно-мышечные заболевания характеризуются нарушением движений и прогрессирующей атрофией мышц. Выделяют: прогрессирующие мышечные дистрофии (миопатии); спинальные амиотрофии;
- 13. Наследственные болезни мышц, мышечные дистрофии и миопатии
- 14. Клиническая классификация прогрессирующих мышечных дистрофий (ПМД) основана на характере распространения мышечных атрофий и парезов – конечностно-поясные,
- 15. Морфологически в мышечных волокнах при миопатиях выявляются дистрофические и некротические изменения, разрастание соединительной ткани, диффузная разнокалиберность
- 16. Биохимически в сыворотке крови увеличено содержание саркоплазматических ферментов – креатинфосфокиназы (КФК), альдолазы, лактатдегидрогеназы и др. Эти
- 17. Наиболее распространенной и злокачественной формой нервно-мышечной патологии детского возраста является прогрессирующая Х-сцепленная псевдогипертрофическая миодистрофия Дюшенна/Беккера
- 18. Первые признаки миодистрофии Дюшенна появляются в возрасте 2-7 лет. При начале ходьбы отмечаются неловкость в движениях,
- 19. Патологический процесс носит восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем
- 21. Характерно лицо «сфинкса», «миопата» - гипертелоризм, недостаточность мимической мускулатуры. Возникают вторичные деформации позвоночника, грудной клетки, ретракции
- 22. Больные миодистрофией Дюшенна сохраняют способность к ходьбе до 10-12-летнего возраста, после чего передвигаются только с помощью
- 23. Миодистрофия Беккера – это более мягкий вариант Х-сцепленной прогрессирующей мышечной дистрофии. В настоящее время убедительно доказано,
- 24. В некоторых случаях дюшенно-подобные миодистрофии наследуются по аутосомно-рецессивному типу. К подобным заболеваниям относится, в частности, врожденная
- 25. Конечностно-поясные миодистрофии – это гетерогенная группа заболеваний с преимущественной локализацией дистрофического процесса в мышцах плечевого и
- 26. Больные начинают испытывать затруднения при беге и ходьбе в 15-20 лет, однако способность к самостоятельному передвижению
- 27. Генетическое разнообразие наследственных конечностно-поясных миодистрофий очень велико. Идентифицированы мутантные гены для четырех аутосомно-рецессивных и десяти аутосомно-доминантных
- 28. При некоторых формах конечностно-поясная миодистрофия сочетается с выраженной патологией других систем, например с буллезным эпидермолизом или
- 29. В самостоятельную клиническую группу традиционно выделяют врожденные непрогрессирующие миопатии. Наиболее частой из них является мерозин-дефицитная миопатия.
- 30. Патологические процессы при некоторых врожденных непрогрессирующих миопатиях обусловлены отложением в миофибриллах гистологически идентифицируемых аномальных образований
- 31. При немалиновой миопатии в мышечных клетках пациентов присутствуют нитеобразные патологические фибриллярные структуры, причиной развития которых является
- 32. Определенные гистологические аномалии выявляются также у больных миотубулярной миопатией и болезнью центрального стержня
- 33. Нерастворимые включения в мышечных клетках характерны и для других миопатий, дебютирующих в более позднем возрасте. Примерами
- 34. В особую группу выделяют медленно прогрессирующие митохондриальные миопатии, клиническая картина которых складывается из слабости мышц, начинающейся
- 35. Наследственные дефекты различных мышечных ферментов являются причиной развития относительно доброкачественных метаболических миопатий, таких как мышечный гликогеноз
- 36. В настоящее время расшифрована молекулярно- генетическая природа многих наследственных форм мышечных дистрофий и миопатий, что позволяет
- 37. Оказалось, что белковые продукты многих генов, связанных с наследственными болезнями мышц, ассоциированы с мембранами мышечных волокон
- 38. Основными функциями подобных белков являются: стабилизация сарколеммы мышечного волокна за счет связывания цитоскелета с внеклеточным матриксом
- 39. К подобным белкам относится дистрофин – стержневидный белок, принадлежащий к спектрин/α-актининовому суперсемейству белков цитоскелета
- 40. Дистрофин-ассоциированный комплекс белков
- 41. N-концевой домен дистрофина связан с цитоскелетом мышечного волокна. Затем идет самый крупный стержневидный домен, обеспечивающий гибкость
- 42. В области цистеин-богатого домена формируются кальциевые каналы и осуществляется связь дистрофина, а значит и цитоскелета мышечного
- 43. Эти белки, в свою очередь, разделяют на два субкомплекса – саркогликановый и дистрогликановый. В области С-концевого
- 44. При миодистрофии Дюшенна/Беккера, также как при аутосомно-рецессивных дюшенно-подобных и некоторых конечностно-поясных миодистрофиях происходит разрушение дистрофин-ассоциированного комплекса
- 45. При миодистрофии Дюшенна/Беккера разрушение дистрофин-ассоциированного комплекса белков происходит за счет мутаций в гене дистрофина. Таким образом
- 46. Разрушение дистрофинового комплекса за счет специфических мутаций в гене DMD является одним из центральных звеньев в
- 47. В 65-70% случаев у больных миодистрофией Дюшенна/Беккера диагностируются протяженные внутригенные делеции, затрагивающие несколько соседних экзонов гена
- 48. Различия заключаются в том, что при миодистрофии Дюшенна делеции сопровождаются сдвигом рамки считывания, и дистрофин у
- 49. В гене DMD идентифицированы также относительно небольшие перестройки и нонсенс-мутации, в то время как миссенс-мутации встречаются
- 50. Молекулярная диагностика делеций в гене DMD проводится с использованием мультиплексной ПЦР, что позволяет во многих семьях
- 52. Разрушение дистрофин-ассоциированного комплекса белков при аутосомно-рецессивных дюшенно-подобных и некоторых конечностно-поясных миодистрофиях происходит за счет мутаций в
- 53. Дистрофин-ассоциированный комплекс белков
- 54. Мутации в генах дистрогликанов не обнаружены. Но при врожденных миодистрофиях, сопровождающихся тяжелой умственной отсталостью, найдены мутации
- 55. Дистрогликанопатии (5 нозологических форм) Миодистрофия врожденная, прогрессирующая с умственной отсталостью, тип Фукуяма, аутосомно-рецессивная Уолкера-Варбурга синдром Миодистрофия
- 56. Некоторые формы дистальных миопатий обусловлены мутациями в гене кавеолина – основного белка кавеоловых мембран сарколеммы мышечного
- 57. Кавеолинопатии (7нозологических форм) Миодистрофия конечностно-поясная, аутосомно-доминантная, 1C миопатия дистальная болезнь волнистых (rippling) мышц Миодистрофия конечностно-поясная, аутосомно-рецессивная,
- 58. Инактивирующие мутации в гене плектина, участвующего в связи цитоскелета мышечного волокна с мембраной, приводят к необычной
- 59. Сарколеммные миопатии (20 нозологических форм) Дистрофинопатии (миодистофия Дюшенна/Беккера) Саркогликанопатии (4 формы конечностно-поясных миодистофий) Дистрогликанопатии (5 форм
- 60. Наследственная недостаточность белков внеклеточного матрикса, взаимодействующих с дистрогликановым комплексом, является причиной развития врожденных аутосомно-рецессивных болезней мышц,
- 61. К матриксным миопатиям относятся также аутосомно-доминантные врожденные миопатии Бетлема и Ульриха. Эти заболевания обусловлены дефектами трех
- 62. Матриксные миопатии (4 нозологические формы) Миодистрофия врожденная, мерозин-дефицитная, аутосомно-рецессивная Миопатия врожденная, интегрин α7 β1D-дефицитная, аутосомно-рецессивная Миопатия
- 63. Нарушения структуры белков ядерной ламины миофибрилл ассоциированы с другой группой мышечных дистрофий, получивших название ламинопатии. Это
- 64. При наиболее распространенной Х-сцепленной форме заболевания мутантным оказывается ген EMD (Xq28) ядерного ламина-ассоциированного белка, получившего название
- 65. Более редкие аутосомные формы миодистрофии Эмери-Дрейфуса обусловлены мутациями в гене LMNA (1q22), кодирующем синтез двух филаментных
- 66. Нарушения структуры белков ядерной ламины миофибрилл – ламинопатии (3 нозологические формы) миодистрофия с контрактурами Эмери-Дрейфуса, Х-сцепленная
- 67. Мутации в гене ламина A/C, приводят к 13 заболеваниям, включающим наряду с мышечными дистрофиями различные варианты
- 68. Хорошо известная клиницистам немалиновая, или нитчатая миопатия также представляет собой генетически гетерогенную группу заболеваний. Данная разновидность
- 69. Миопатия немалиновая небулин – интегральный компонент тонких (актиновых) и толстых (миозиновых) филамент саркомера тропомиозин 2 –
- 70. Миотилиновые, титиновые и телетониновые конечностно-поясные миопатии 1А: миотилин – гигантский саркомерный белок, содержащий несколько Ig-подобных доменов,
- 71. Саркомерные миопатии (15 нозологических форм) Немалиновая миопатия (6 нозологических форм) Миотилиновые, титиновые и телетониновые конечностно-поясные миопатии
- 72. При целом ряде миопатий причиной дистрофических процессов является накопление в цитоплазме и/или в ядрах мышечных клеток
- 73. Определенные гистологические аномалии характерны для пациентов с болезнью центрального стержня и миотубулярной миопатией. В обеих случаях
- 74. В первом случае – это рецептор 1 рионадина – кальций высвобождающего канала саркоплазматического ретикулума скелетных мышц,
- 75. Для ряда миопатий, дебютирующих в позднем возрасте и сочетающихся с нарушениями сердечной проводимости, аритмиями и рестриктивной
- 76. Причиной развития патологических процессов в данных случаях являются нарушения конформации определенных мышечных белков, то есть это
- 77. Конформационные болезни мышц десминовые миопатии, обусловленные накоплением цитоплазматических филаментных включений миопатии с инклюзионными тельцами, при которых
- 78. Биохимическая классификация миодистрофий и миопатий (60 нозологических форм) Сарколеммные миопатии (20) Матриксные миопатии (4) Ламинопатии (3)
- 79. Таким образом, различные нарушения могут приводить к развитию сходных патологических процессов в мышцах. При постановке дифференциального
- 80. Однако классификации наследственных болезней мышц, основанные на морфологических и патогенетических критериях, далеко не всегда совпадают. Это
- 81. Миодистрофии конечностно-поясные Доминантные 1А - миотилин 1B - ламин A/C 1C - кавеолин-3 Рецессивные 2A -
- 82. Лице-лопаточно-плечевая мышечная дистрофия Ландузи-Дежерина – третье по частоте аутосомно-доминантное заболевание мышц. Первые признаки заболевания обычно появляются
- 83. Преимущественно поражается мускулатура лица, плечевого пояса и проксимальных отделов верхних конечностей. Слабость мускулатуры лица проявляется неполным
- 84. В области локализации мутантного локуса FSHD1 (4q35-qter), ответственного за развитие заболевания, расположен высоко полиморфный макросателлитный повтор
- 85. В норме количество копий этого повтора варьирует от 11 до 100. У больных наблюдаются гетерозиготные делеции
- 86. Предполагается, что мутации, вызывающие лице-лопаточно-плечевую миодистрофию, нарушают не структуру или функцию какого-то специфического гена, но транскрипционный
- 87. Сложные эпигенетические механизмы вовлечены в реализацию этого нарушения. Молекулярная диагностика заболевания основана на определении величины D4Z4-повтора
- 88. Спинальные амиотрофии – это гетерогенная группа наследственных заболеваний, обусловленных прогрессирующим разрушением мотонейронов передних рогов спинного мозга
- 89. Наиболее распространенной аутосомно-рецессивной формой поражения периферического двигательного неврона является проксимальная спинальная мышечная атрофия (СМА). Частота заболевания
- 90. Основными клиническими проявлениями СМА являются слабость и гипотония мышц, вялые симметричные парезы всей поперечно-полосатой мускулатуры, угнетение
- 91. СМА делят на 3 формы: болезнь Верднига-Гоффмана, острая детская СМА I, с дебютом до 6 месяцев,
- 92. Болезнь Верднига-Гоффмана может проявляться уже во внутриутробном периоде недостаточно активным шевелением плода, при рождении – генерализованной
- 93. Развиваются вялые, преимущественно проксимальные парезы конечностей, глубокие рефлексы отсутствуют. Обычно больные погибают в течение первого года
- 94. При СМА II ребёнок может научиться сидеть и даже ходить с поддержкой. При этом походка раскачивающаяся,
- 95. Болезнь Кугельберга-Веландер характеризуется более медленным течением сходных патологических процессов на протяжении десятилетий. Продолжительность жизни при СМА
- 96. Все клинические типы СМА обусловлены мутациями в гене SMN1 (5q13.2), кодирующем белок выживания двигательных нейронов –
- 98. При инактивации Smn-белка периферические двигательные нейроны теряют способность контролировать образование мРНК, в результате чего нарушается синтез
- 99. В непосредственной близости от гена SMN1 идентифицирован его гомолог, получивший название SMN2. У разных индивидуумов ген
- 100. Характер экспрессии двух гомологичных генов SMN1 и SMN2 в специализированных тканях организма одинаков, но их продукты
- 101. Поэтому присутствие у больных СМА трех и более дополнительных копий гена SMN2 достоверно коррелирует с более
- 102. Одна из главных стратегий лечения СМА направлена на повышение активности гена SMN2. Первые результаты применения вальпроевой
- 103. Боковой амиотрофический склероз (БАС) характеризуется своеобразным сочетанием поражения периферического и центрального двигательных невронов. Клиническая картина складывается
- 104. БАС начинается в среднем возрасте и в дальнейшем прогрессирует. В 10% случаев заболевание носит семейный характер
- 105. Для наследственных форм БАС характерна большая генетическая гетерогенность, хотя в 20% случаев у больных обнаруживаются мутации
- 106. Наследственные полиневропатии
- 107. Наследственные полиневропатии составляют до 60-70% всех хронических полиневропатий и представляют собой весьма гетерогенную группу. Наиболее распространёнными
- 108. Клинически характеризуются прогрессирующей слабостью и атрофией дистальной (преимущественно перонеальной) мускулатуры; расстройствами чувствительности по полиневритическому типу; деформацией
- 109. Патогенетически моторно-сенсорная невропатия делится на 2 основных типа: демиелинизирующая – миелопатия и аксональная– аксонопатия. Для первых
- 110. Для второго типа характерно первичное поражение аксонов, нормальная скорость проведения импульса, морфологически сохранность структуры миелина
- 111. Из сравнительно редких синдромов, отличающихся от классического фенотипа Шарко-Мари-Тута следует отметить синдром Дежерина-Сотта, основными клиническими проявлениями
- 112. Для моторно-сенсорных полинейропатий характерна огромная генетическая гетерогенность. В настоящее время идентифицированы более 20 генов, мутации в
- 113. Наиболее частыми являются аутосомно-доминантные демиелинизирующие полинейропатии, связанные с нарушением синтеза миелина периферических нервов
- 114. Тип 1А, обусловлен гиперпродукцией интегрального белка компактного миелина периферической нервной системы – pmp22. Гиперпродукция миелинового белка
- 115. При типе 1B дефектным оказывается структурный белок периферического миелина P(0), кодируемый геном MPZ (1q23.3). P(0) составляет
- 116. Относительно редкие аксональные формы аутосомно-доминантной болезни Шарко-Мари-Тута, промежуточные и аутосомно-рецессивные формы еще более гетерогенны по количеству
- 117. Синдром Дежерина-Сотта также генетически гетерогенен, но не является самостоятельной формой, а представляет собой аллельные варианты различных
- 118. Наследственные миотонии и миоплегии (нервно-мышечные каналопатии)
- 119. Ионные каналы участвуют в поддержании мембранного потенциала, регуляции объема и модуляции электрической возбудимости во многих типах
- 120. Дефектная работа ионных каналов в кардиомиоцитах ассоциирована с нарушениям сердечного ритма. Мутации в генах некоторых нейрональных
- 121. Феномен миотонии заключается во внезапном тоническом спазме мышцы, возникающем вслед за произвольным её сокращением. Он может
- 122. Известны две клинические формы: аутосомно-доминантная врождённая миотония Томсона и аутосомно-рецессивная генерализованная миотония Беккера. Характерной особенностью миотонии
- 123. Обе формы обусловлены мутациями в гене хлорного канала скелетных мышц – CLC1 (7q34). Известно, что ионы
- 124. Наследственная пароксизмальная миоплегия характеризуется приступами резкой слабости, вплоть до полного паралича рук и ног. Выделяют 3
- 125. Аутосомно-доминантная гипокалиемическая форма, или болезнь Шахновича-Вестфаля встречается наиболее часто. Приступы появляются с детства, от единичных до
- 126. Краниальная мускулатура, как правило, не страдает, сознание сохранено. Длительность приступа от 30 минут до 72 часов,
- 127. Гиперкалиемическая форма пароксизмальной миоплегии, или болезнь Гармстропа отличается слабостью мимической и артикуляционной мускулатуры, приступ может провоцироваться
- 128. При гипокалиемическом параличе, обусловленном мутациями в гене CACNA1S (1q31-32), дефектным оказывается кальциевый канал L-типа. При этом
- 129. Мутации в гене SCN4 (17q23.1), кодирующем альфа-4-субъединицу потенциал-зависимого натриевого канала, обнаруживаются при гиперкалиемическом периодическом параличе и
- 130. В результате дефекта натриевого канала происходит усиление тока иона в клетку, генерация потенциала действия и деполяризация
- 131. Наследственные миастении
- 132. В разделе наследственных нейромышечных заболеваний традиционно рассматривают миастению, или болезнь Эрба. Это многофакторное аутоиммунное заболевание, относящееся
- 133. Миастения клинически характеризуется нарастающей при выполнении движений мышечной слабостью, которая может достигать степени паралича. После отдыха
- 134. Различают миастению генерализованную и локальную с поражением глазодвигательных нервов (глазная форма) и мышц гортани, глотки, языка
- 135. Опасны миастенические кризы, когда внезапно развиваются генерализованная общая слабость и выраженные бульбарные расстройства, нарушения дыхания, требующие
- 136. Как и для многих других аутоиммунных заболеваний, генетическими факторами риска миастении являются полиморфные аллели HLA-генов главного
- 137. В патогенезе аутоиммунной реакции основную роль играет тимус. При патологии тимоцитов (тимома, воспаление и др.) их
- 138. Патогенетическая терапия: антихолинэстеразные препараты (прозерин, оксазил, каллимин), курсы стероидной терапии. При выявлении опухоли вилочковой железы её
- 139. Редкие врожденные миастенические синдромы – это моногенные формы миастении. Их классифицируют по месту трансмиссионного дефекта: пресинаптические
- 140. Пресинаптический миастенически синдром, сопровождающийся эпизодической атаксией, обусловлен мутациями в гене CHAT (10q11.23), кодирующем холинацетилтрансферазу – биосинтетический
- 141. Постсинаптический синдром по характеру кинетического дефекта ацетилхолинового рецептора делится на два варианта — с быстрым и
- 142. И в том, и в другом случае мутантными оказываются гены различных субъединиц ацетилхолинового рецептора: альфа (CHRNA1,
- 143. При этом причиной развития варианта с быстрым потоком являются инактивирующие мутации, и заболевание в этом случае
- 144. При варианте с медленным потоком мутации в генах различных субъединиц ацетилхолинового рецептора обладают доминантно-негативным эффектом, а
- 145. В некоторых случаях у больных с врожденным миастеническим синдромом обнаруживают мутации в гене RAPSN (11p11.2), продукт
- 146. Синаптический вариант врожденного миастенического синдрома обусловлен недостаточностью синаптической ацетилхолинэстеразы, причиной развития которой являются инактивирующие рецессивные мутации
- 147. При нарушении структуры коллагена Q не происходит встраивания ацетилхолинэстеразы в базальную пластинку синаптической щели
- 149. Скачать презентацию

Понимание ведущей роли генетических факторов в этиологии и патогенезе многих болезней
Понимание ведущей роли генетических факторов в этиологии и патогенезе многих болезней

Конец XX века и начало нового тысячелетия ознаменовался бурным развитием молекулярной
Конец XX века и начало нового тысячелетия ознаменовался бурным развитием молекулярной

В 1990-2000 годы,
получившие название
«Десятилетие мозга»,
фактически произошла смена
В 1990-2000 годы, получившие название «Десятилетие мозга», фактически произошла смена

Идентификация мутантных генов и первичных биохимических нарушений, ответственных за развитие многих
Идентификация мутантных генов и первичных биохимических нарушений, ответственных за развитие многих

На этой основе были сформированы новые неизвестные ранее классы заболеваний, такие
На этой основе были сформированы новые неизвестные ранее классы заболеваний, такие

В основе болезней экспансии,
лежат динамические мутации, обусловленные нестабильностью расположенных в
В основе болезней экспансии, лежат динамические мутации, обусловленные нестабильностью расположенных в

Патогенетический механизм нейродегенеративных болезней, обусловленных экспансией CAG-или GCG-повторов, расположенных в кодирующих
Патогенетический механизм нейродегенеративных болезней, обусловленных экспансией CAG-или GCG-повторов, расположенных в кодирующих

Подобные болезни получили название конформационных.
К ним относятся некоторые миопатии, синдромы
Подобные болезни получили название конформационных. К ним относятся некоторые миопатии, синдромы

При наследственных заболеваниях с пароксизмами, гипер- или гипо-возбудимостью вовлеченных в патологический
При наследственных заболеваниях с пароксизмами, гипер- или гипо-возбудимостью вовлеченных в патологический

К каналопатиям относятся миотонии, периодические параличи, наследственные формы абсанс эпилепсии и
К каналопатиям относятся миотонии, периодические параличи, наследственные формы абсанс эпилепсии и

Нервно-мышечные заболевания характеризуются нарушением движений и прогрессирующей атрофией мышц.
Выделяют: прогрессирующие
Нервно-мышечные заболевания характеризуются нарушением движений и прогрессирующей атрофией мышц. Выделяют: прогрессирующие

Наследственные болезни мышц, мышечные дистрофии и миопатии
Наследственные болезни мышц, мышечные дистрофии и миопатии

Клиническая классификация прогрессирующих мышечных дистрофий (ПМД) основана на характере распространения мышечных
Клиническая классификация прогрессирующих мышечных дистрофий (ПМД) основана на характере распространения мышечных

Морфологически в мышечных волокнах при миопатиях выявляются дистрофические и некротические изменения,
Морфологически в мышечных волокнах при миопатиях выявляются дистрофические и некротические изменения,

Биохимически в сыворотке крови увеличено содержание саркоплазматических ферментов – креатинфосфокиназы (КФК),
Биохимически в сыворотке крови увеличено содержание саркоплазматических ферментов – креатинфосфокиназы (КФК),

Наиболее распространенной и злокачественной формой нервно-мышечной патологии детского возраста является
прогрессирующая
Наиболее распространенной и злокачественной формой нервно-мышечной патологии детского возраста является прогрессирующая

Первые признаки миодистрофии Дюшенна появляются
в возрасте 2-7 лет.
При начале
Первые признаки миодистрофии Дюшенна появляются в возрасте 2-7 лет. При начале

Патологический процесс носит восходящий характер. Первыми поражаются мышцы тазового пояса и
Патологический процесс носит восходящий характер. Первыми поражаются мышцы тазового пояса и
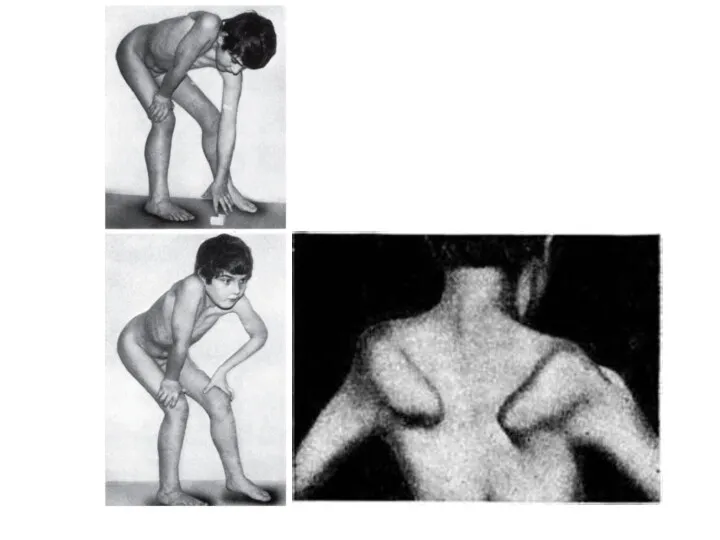

Характерно лицо «сфинкса», «миопата» - гипертелоризм, недостаточность мимической мускулатуры.
Возникают вторичные
Характерно лицо «сфинкса», «миопата» - гипертелоризм, недостаточность мимической мускулатуры. Возникают вторичные

Больные миодистрофией Дюшенна сохраняют способность к ходьбе до 10-12-летнего возраста, после
Больные миодистрофией Дюшенна сохраняют способность к ходьбе до 10-12-летнего возраста, после

Миодистрофия Беккера – это более мягкий вариант Х-сцепленной прогрессирующей мышечной дистрофии.
Миодистрофия Беккера – это более мягкий вариант Х-сцепленной прогрессирующей мышечной дистрофии.

В некоторых случаях дюшенно-подобные миодистрофии наследуются по аутосомно-рецессивному типу.
К подобным
В некоторых случаях дюшенно-подобные миодистрофии наследуются по аутосомно-рецессивному типу. К подобным

Конечностно-поясные миодистрофии –
это гетерогенная группа заболеваний с преимущественной локализацией дистрофического
Конечностно-поясные миодистрофии – это гетерогенная группа заболеваний с преимущественной локализацией дистрофического

Больные начинают испытывать затруднения при беге и ходьбе в 15-20 лет,
Больные начинают испытывать затруднения при беге и ходьбе в 15-20 лет,

Генетическое разнообразие наследственных конечностно-поясных миодистрофий очень велико.
Идентифицированы мутантные гены для
Генетическое разнообразие наследственных конечностно-поясных миодистрофий очень велико. Идентифицированы мутантные гены для

При некоторых формах конечностно-поясная миодистрофия сочетается с выраженной патологией других систем,
При некоторых формах конечностно-поясная миодистрофия сочетается с выраженной патологией других систем,

В самостоятельную клиническую группу традиционно выделяют врожденные непрогрессирующие миопатии.
Наиболее частой
В самостоятельную клиническую группу традиционно выделяют врожденные непрогрессирующие миопатии. Наиболее частой

Патологические процессы при некоторых врожденных непрогрессирующих миопатиях обусловлены отложением в миофибриллах
Патологические процессы при некоторых врожденных непрогрессирующих миопатиях обусловлены отложением в миофибриллах

При немалиновой миопатии в мышечных клетках пациентов присутствуют нитеобразные патологические фибриллярные
При немалиновой миопатии в мышечных клетках пациентов присутствуют нитеобразные патологические фибриллярные

Определенные гистологические аномалии выявляются также у больных миотубулярной миопатией и болезнью
Определенные гистологические аномалии выявляются также у больных миотубулярной миопатией и болезнью

Нерастворимые включения в мышечных клетках характерны и для других миопатий, дебютирующих
Нерастворимые включения в мышечных клетках характерны и для других миопатий, дебютирующих

В особую группу выделяют медленно прогрессирующие митохондриальные миопатии,
клиническая картина которых
В особую группу выделяют медленно прогрессирующие митохондриальные миопатии, клиническая картина которых

Наследственные дефекты различных мышечных ферментов являются причиной развития относительно доброкачественных метаболических
Наследственные дефекты различных мышечных ферментов являются причиной развития относительно доброкачественных метаболических

В настоящее время расшифрована молекулярно- генетическая природа многих наследственных форм мышечных
В настоящее время расшифрована молекулярно- генетическая природа многих наследственных форм мышечных

Оказалось, что белковые продукты многих генов, связанных с наследственными болезнями мышц,
Оказалось, что белковые продукты многих генов, связанных с наследственными болезнями мышц,

Основными функциями подобных белков являются:
стабилизация сарколеммы мышечного волокна за счет
Основными функциями подобных белков являются:
стабилизация сарколеммы мышечного волокна за счет

К подобным белкам относится дистрофин –
стержневидный белок, принадлежащий к
спектрин/α-актининовому
К подобным белкам относится дистрофин – стержневидный белок, принадлежащий к спектрин/α-актининовому
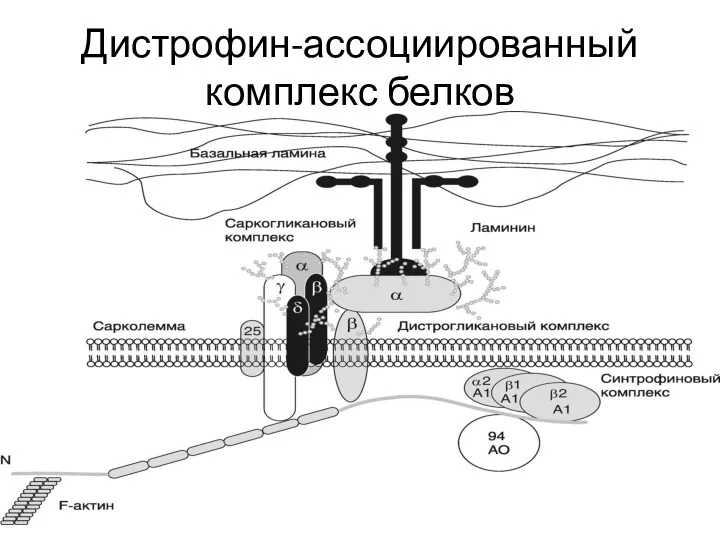
Дистрофин-ассоциированный комплекс белков
Дистрофин-ассоциированный комплекс белков

N-концевой домен дистрофина связан с цитоскелетом мышечного волокна.
Затем идет самый
N-концевой домен дистрофина связан с цитоскелетом мышечного волокна. Затем идет самый

В области цистеин-богатого домена формируются кальциевые каналы и осуществляется связь дистрофина,
В области цистеин-богатого домена формируются кальциевые каналы и осуществляется связь дистрофина,

Эти белки, в свою очередь, разделяют на два субкомплекса – саркогликановый
Эти белки, в свою очередь, разделяют на два субкомплекса – саркогликановый

При миодистрофии Дюшенна/Беккера, также как при аутосомно-рецессивных дюшенно-подобных и некоторых конечностно-поясных
При миодистрофии Дюшенна/Беккера, также как при аутосомно-рецессивных дюшенно-подобных и некоторых конечностно-поясных

При миодистрофии Дюшенна/Беккера разрушение
дистрофин-ассоциированного комплекса белков происходит за счет мутаций
При миодистрофии Дюшенна/Беккера разрушение дистрофин-ассоциированного комплекса белков происходит за счет мутаций

Разрушение дистрофинового комплекса за счет специфических мутаций в гене DMD является
Разрушение дистрофинового комплекса за счет специфических мутаций в гене DMD является

В 65-70% случаев у больных миодистрофией Дюшенна/Беккера диагностируются протяженные внутригенные делеции,
В 65-70% случаев у больных миодистрофией Дюшенна/Беккера диагностируются протяженные внутригенные делеции,

Различия заключаются в том, что при миодистрофии Дюшенна делеции сопровождаются сдвигом
Различия заключаются в том, что при миодистрофии Дюшенна делеции сопровождаются сдвигом

В гене DMD идентифицированы также относительно небольшие перестройки и нонсенс-мутации, в
В гене DMD идентифицированы также относительно небольшие перестройки и нонсенс-мутации, в

Молекулярная диагностика делеций в гене DMD проводится с использованием мультиплексной ПЦР,
Молекулярная диагностика делеций в гене DMD проводится с использованием мультиплексной ПЦР,
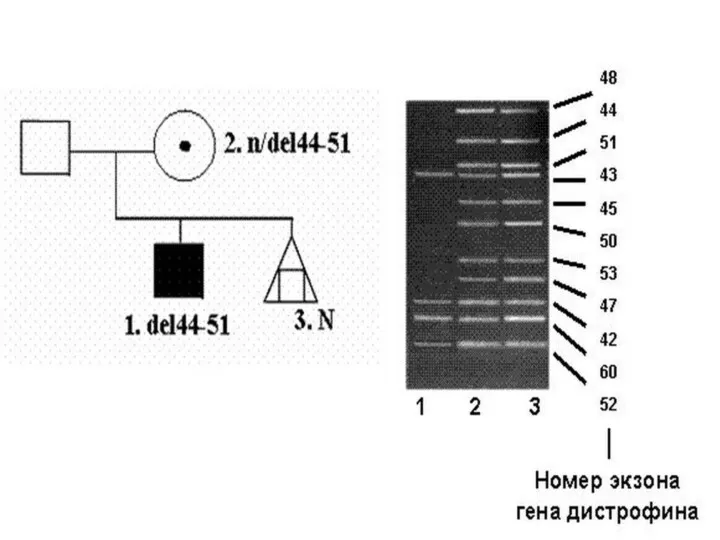

Разрушение дистрофин-ассоциированного комплекса белков при аутосомно-рецессивных дюшенно-подобных и некоторых конечностно-поясных миодистрофиях
Разрушение дистрофин-ассоциированного комплекса белков при аутосомно-рецессивных дюшенно-подобных и некоторых конечностно-поясных миодистрофиях
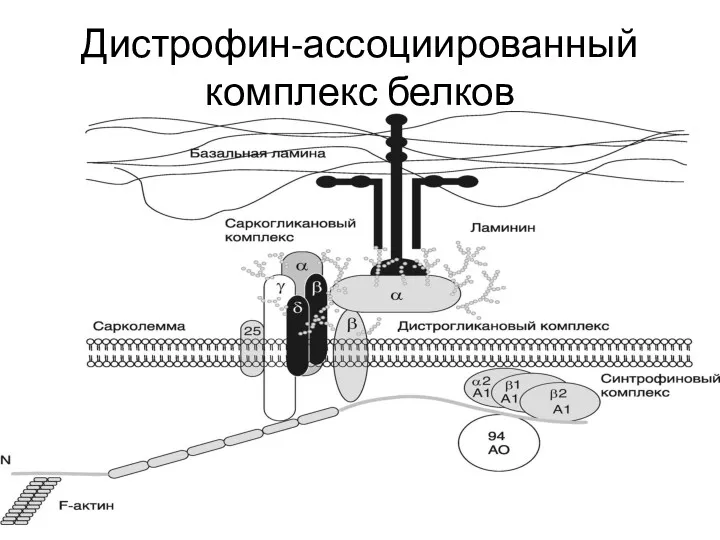
Дистрофин-ассоциированный комплекс белков
Дистрофин-ассоциированный комплекс белков

Мутации в генах дистрогликанов не обнаружены.
Но при врожденных миодистрофиях, сопровождающихся
Мутации в генах дистрогликанов не обнаружены. Но при врожденных миодистрофиях, сопровождающихся

Дистрогликанопатии
(5 нозологических форм)
Миодистрофия врожденная, прогрессирующая с умственной отсталостью, тип
Дистрогликанопатии
(5 нозологических форм)
Миодистрофия врожденная, прогрессирующая с умственной отсталостью, тип

Некоторые формы дистальных миопатий обусловлены мутациями в гене кавеолина – основного
Некоторые формы дистальных миопатий обусловлены мутациями в гене кавеолина – основного

Кавеолинопатии
(7нозологических форм)
Миодистрофия конечностно-поясная, аутосомно-доминантная, 1C
миопатия дистальная
болезнь волнистых
Кавеолинопатии
(7нозологических форм)
Миодистрофия конечностно-поясная, аутосомно-доминантная, 1C
миопатия дистальная
болезнь волнистых

Инактивирующие мутации в гене плектина, участвующего в связи цитоскелета мышечного волокна
Инактивирующие мутации в гене плектина, участвующего в связи цитоскелета мышечного волокна

Сарколеммные миопатии
(20 нозологических форм)
Дистрофинопатии (миодистофия Дюшенна/Беккера)
Саркогликанопатии (4 формы конечностно-поясных миодистофий)
Дистрогликанопатии (5
Сарколеммные миопатии
(20 нозологических форм)
Дистрофинопатии (миодистофия Дюшенна/Беккера)
Саркогликанопатии (4 формы конечностно-поясных миодистофий)
Дистрогликанопатии (5

Наследственная недостаточность белков внеклеточного матрикса, взаимодействующих с дистрогликановым комплексом, является причиной
Наследственная недостаточность белков внеклеточного матрикса, взаимодействующих с дистрогликановым комплексом, является причиной

К матриксным миопатиям относятся также аутосомно-доминантные врожденные миопатии
Бетлема и Ульриха.
К матриксным миопатиям относятся также аутосомно-доминантные врожденные миопатии Бетлема и Ульриха.

Матриксные миопатии
(4 нозологические формы)
Миодистрофия врожденная, мерозин-дефицитная, аутосомно-рецессивная
Миопатия врожденная, интегрин
Матриксные миопатии
(4 нозологические формы)
Миодистрофия врожденная, мерозин-дефицитная, аутосомно-рецессивная
Миопатия врожденная, интегрин

Нарушения структуры белков ядерной ламины миофибрилл ассоциированы с другой группой мышечных
Нарушения структуры белков ядерной ламины миофибрилл ассоциированы с другой группой мышечных

При наиболее распространенной Х-сцепленной форме заболевания мутантным оказывается ген EMD (Xq28)
При наиболее распространенной Х-сцепленной форме заболевания мутантным оказывается ген EMD (Xq28)

Более редкие аутосомные формы миодистрофии Эмери-Дрейфуса обусловлены мутациями в гене LMNA
Более редкие аутосомные формы миодистрофии Эмери-Дрейфуса обусловлены мутациями в гене LMNA

Нарушения структуры белков ядерной ламины миофибрилл – ламинопатии
(3 нозологические формы)
Нарушения структуры белков ядерной ламины миофибрилл – ламинопатии (3 нозологические формы)

Мутации в гене ламина A/C, приводят к 13 заболеваниям, включающим наряду
Мутации в гене ламина A/C, приводят к 13 заболеваниям, включающим наряду

Хорошо известная клиницистам немалиновая, или нитчатая миопатия также представляет собой генетически
Хорошо известная клиницистам немалиновая, или нитчатая миопатия также представляет собой генетически

Миопатия немалиновая
небулин – интегральный компонент тонких (актиновых) и толстых (миозиновых)
Миопатия немалиновая
небулин – интегральный компонент тонких (актиновых) и толстых (миозиновых)

Миотилиновые, титиновые и телетониновые конечностно-поясные миопатии
1А: миотилин – гигантский саркомерный белок,
Миотилиновые, титиновые и телетониновые конечностно-поясные миопатии
1А: миотилин – гигантский саркомерный белок,

Саркомерные миопатии
(15 нозологических форм)
Немалиновая миопатия
(6 нозологических форм)
Саркомерные миопатии
(15 нозологических форм)
Немалиновая миопатия
(6 нозологических форм)

При целом ряде миопатий причиной дистрофических процессов является накопление в цитоплазме
При целом ряде миопатий причиной дистрофических процессов является накопление в цитоплазме

Определенные гистологические аномалии характерны для пациентов с болезнью центрального стержня и
Определенные гистологические аномалии характерны для пациентов с болезнью центрального стержня и

В первом случае – это рецептор 1 рионадина – кальций высвобождающего
В первом случае – это рецептор 1 рионадина – кальций высвобождающего

Для ряда миопатий, дебютирующих в позднем возрасте и сочетающихся с нарушениями
Для ряда миопатий, дебютирующих в позднем возрасте и сочетающихся с нарушениями

Причиной развития патологических процессов в данных случаях являются нарушения конформации определенных
Причиной развития патологических процессов в данных случаях являются нарушения конформации определенных

Конформационные болезни мышц
десминовые миопатии, обусловленные накоплением цитоплазматических филаментных включений
миопатии с
Конформационные болезни мышц
десминовые миопатии, обусловленные накоплением цитоплазматических филаментных включений
миопатии с

Биохимическая классификация миодистрофий и миопатий
(60 нозологических форм)
Сарколеммные миопатии (20)
Матриксные миопатии (4)
Биохимическая классификация миодистрофий и миопатий
(60 нозологических форм)
Сарколеммные миопатии (20)
Матриксные миопатии (4)

Таким образом, различные нарушения могут приводить к развитию сходных патологических процессов
Таким образом, различные нарушения могут приводить к развитию сходных патологических процессов

Однако классификации наследственных болезней мышц, основанные на морфологических и патогенетических критериях,
Однако классификации наследственных болезней мышц, основанные на морфологических и патогенетических критериях,

Миодистрофии конечностно-поясные
Доминантные
1А - миотилин
1B - ламин A/C
Миодистрофии конечностно-поясные
Доминантные
1А - миотилин
1B - ламин A/C

Лице-лопаточно-плечевая мышечная дистрофия Ландузи-Дежерина – третье по частоте аутосомно-доминантное заболевание мышц.
Лице-лопаточно-плечевая мышечная дистрофия Ландузи-Дежерина – третье по частоте аутосомно-доминантное заболевание мышц.

Преимущественно поражается мускулатура лица, плечевого пояса и проксимальных отделов верхних конечностей.
Преимущественно поражается мускулатура лица, плечевого пояса и проксимальных отделов верхних конечностей.

В области локализации мутантного локуса FSHD1 (4q35-qter), ответственного за развитие заболевания,
В области локализации мутантного локуса FSHD1 (4q35-qter), ответственного за развитие заболевания,

В норме количество копий этого повтора варьирует
от 11 до 100.
В норме количество копий этого повтора варьирует от 11 до 100.

Предполагается, что мутации, вызывающие лице-лопаточно-плечевую миодистрофию, нарушают не структуру или функцию
Предполагается, что мутации, вызывающие лице-лопаточно-плечевую миодистрофию, нарушают не структуру или функцию

Сложные эпигенетические механизмы вовлечены в реализацию этого нарушения. Молекулярная диагностика заболевания
Сложные эпигенетические механизмы вовлечены в реализацию этого нарушения. Молекулярная диагностика заболевания

Спинальные амиотрофии – это гетерогенная группа наследственных заболеваний, обусловленных прогрессирующим разрушением
Спинальные амиотрофии – это гетерогенная группа наследственных заболеваний, обусловленных прогрессирующим разрушением

Наиболее распространенной аутосомно-рецессивной формой поражения периферического двигательного неврона является проксимальная спинальная
Наиболее распространенной аутосомно-рецессивной формой поражения периферического двигательного неврона является проксимальная спинальная

Основными клиническими проявлениями СМА являются слабость и гипотония мышц, вялые симметричные
Основными клиническими проявлениями СМА являются слабость и гипотония мышц, вялые симметричные

СМА делят на 3 формы: болезнь Верднига-Гоффмана, острая детская СМА I,
СМА делят на 3 формы: болезнь Верднига-Гоффмана, острая детская СМА I,

Болезнь Верднига-Гоффмана может проявляться уже во внутриутробном периоде недостаточно активным шевелением
Болезнь Верднига-Гоффмана может проявляться уже во внутриутробном периоде недостаточно активным шевелением

Развиваются вялые, преимущественно проксимальные парезы конечностей, глубокие рефлексы отсутствуют.
Обычно больные
Развиваются вялые, преимущественно проксимальные парезы конечностей, глубокие рефлексы отсутствуют. Обычно больные

При СМА II ребёнок может научиться сидеть и даже ходить с
При СМА II ребёнок может научиться сидеть и даже ходить с

Болезнь Кугельберга-Веландер характеризуется более медленным течением сходных патологических процессов на протяжении
Болезнь Кугельберга-Веландер характеризуется более медленным течением сходных патологических процессов на протяжении

Все клинические типы СМА обусловлены мутациями в гене SMN1 (5q13.2), кодирующем
Все клинические типы СМА обусловлены мутациями в гене SMN1 (5q13.2), кодирующем
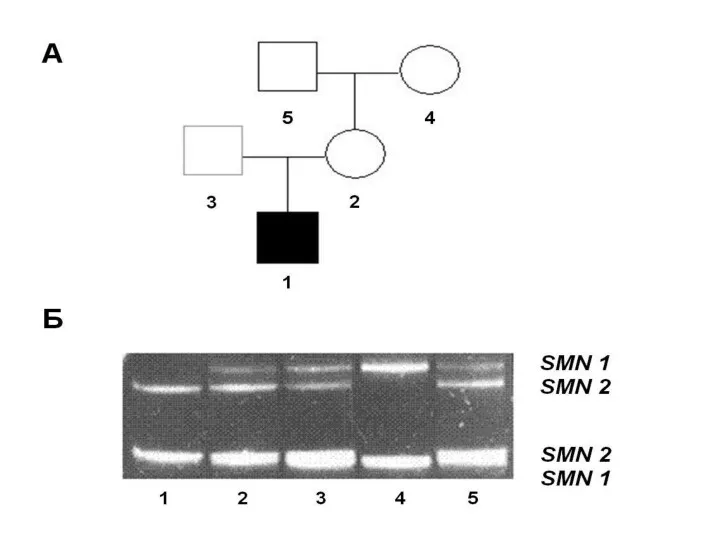

При инактивации Smn-белка периферические двигательные нейроны теряют способность контролировать образование мРНК,
При инактивации Smn-белка периферические двигательные нейроны теряют способность контролировать образование мРНК,

В непосредственной близости от гена SMN1 идентифицирован его гомолог, получивший название
В непосредственной близости от гена SMN1 идентифицирован его гомолог, получивший название

Характер экспрессии двух гомологичных генов SMN1 и SMN2 в специализированных тканях
Характер экспрессии двух гомологичных генов SMN1 и SMN2 в специализированных тканях

Поэтому присутствие у больных СМА трех и более дополнительных копий гена
Поэтому присутствие у больных СМА трех и более дополнительных копий гена

Одна из главных стратегий лечения СМА направлена на повышение активности гена
Одна из главных стратегий лечения СМА направлена на повышение активности гена

Боковой амиотрофический склероз (БАС) характеризуется своеобразным сочетанием поражения периферического и центрального
Боковой амиотрофический склероз (БАС) характеризуется своеобразным сочетанием поражения периферического и центрального

БАС начинается в среднем возрасте и в дальнейшем прогрессирует. В 10%
БАС начинается в среднем возрасте и в дальнейшем прогрессирует. В 10%

Для наследственных форм БАС характерна большая генетическая гетерогенность, хотя в 20%
Для наследственных форм БАС характерна большая генетическая гетерогенность, хотя в 20%

Наследственные полиневропатии
Наследственные полиневропатии

Наследственные
полиневропатии составляют до 60-70% всех хронических полиневропатий и представляют собой
Наследственные полиневропатии составляют до 60-70% всех хронических полиневропатий и представляют собой

Клинически характеризуются прогрессирующей слабостью и атрофией дистальной (преимущественно перонеальной) мускулатуры; расстройствами
Клинически характеризуются прогрессирующей слабостью и атрофией дистальной (преимущественно перонеальной) мускулатуры; расстройствами

Патогенетически моторно-сенсорная невропатия делится на 2 основных типа: демиелинизирующая – миелопатия
Патогенетически моторно-сенсорная невропатия делится на 2 основных типа: демиелинизирующая – миелопатия

Для второго типа характерно первичное поражение аксонов, нормальная скорость проведения импульса,
Для второго типа характерно первичное поражение аксонов, нормальная скорость проведения импульса,

Из сравнительно редких синдромов, отличающихся от классического фенотипа Шарко-Мари-Тута следует отметить
Из сравнительно редких синдромов, отличающихся от классического фенотипа Шарко-Мари-Тута следует отметить

Для моторно-сенсорных полинейропатий характерна огромная генетическая гетерогенность. В настоящее время идентифицированы
Для моторно-сенсорных полинейропатий характерна огромная генетическая гетерогенность. В настоящее время идентифицированы

Наиболее частыми являются аутосомно-доминантные демиелинизирующие полинейропатии, связанные с нарушением синтеза миелина
Наиболее частыми являются аутосомно-доминантные демиелинизирующие полинейропатии, связанные с нарушением синтеза миелина

Тип 1А, обусловлен гиперпродукцией интегрального белка компактного миелина периферической нервной системы
Тип 1А, обусловлен гиперпродукцией интегрального белка компактного миелина периферической нервной системы

При типе 1B дефектным оказывается структурный белок периферического миелина P(0), кодируемый
При типе 1B дефектным оказывается структурный белок периферического миелина P(0), кодируемый

Относительно редкие аксональные формы аутосомно-доминантной болезни Шарко-Мари-Тута, промежуточные и аутосомно-рецессивные формы
Относительно редкие аксональные формы аутосомно-доминантной болезни Шарко-Мари-Тута, промежуточные и аутосомно-рецессивные формы

Синдром Дежерина-Сотта также генетически гетерогенен, но не является самостоятельной формой, а
Синдром Дежерина-Сотта также генетически гетерогенен, но не является самостоятельной формой, а

Наследственные миотонии и миоплегии (нервно-мышечные каналопатии)
Наследственные миотонии и миоплегии (нервно-мышечные каналопатии)

Ионные каналы участвуют в поддержании мембранного потенциала, регуляции объема и модуляции
Ионные каналы участвуют в поддержании мембранного потенциала, регуляции объема и модуляции

Дефектная работа ионных каналов в кардиомиоцитах ассоциирована с нарушениям сердечного ритма.
Дефектная работа ионных каналов в кардиомиоцитах ассоциирована с нарушениям сердечного ритма.

Феномен миотонии заключается во внезапном тоническом спазме мышцы, возникающем вслед за
Феномен миотонии заключается во внезапном тоническом спазме мышцы, возникающем вслед за

Известны две клинические формы: аутосомно-доминантная врождённая миотония Томсона и аутосомно-рецессивная генерализованная
Известны две клинические формы: аутосомно-доминантная врождённая миотония Томсона и аутосомно-рецессивная генерализованная

Обе формы обусловлены мутациями в гене
хлорного канала
скелетных мышц –
Обе формы обусловлены мутациями в гене хлорного канала скелетных мышц –

Наследственная пароксизмальная миоплегия характеризуется приступами резкой слабости, вплоть до полного паралича
Наследственная пароксизмальная миоплегия характеризуется приступами резкой слабости, вплоть до полного паралича

Аутосомно-доминантная гипокалиемическая форма, или болезнь Шахновича-Вестфаля встречается наиболее часто. Приступы появляются
Аутосомно-доминантная гипокалиемическая форма, или болезнь Шахновича-Вестфаля встречается наиболее часто. Приступы появляются

Краниальная мускулатура, как правило, не страдает, сознание сохранено. Длительность приступа от
Краниальная мускулатура, как правило, не страдает, сознание сохранено. Длительность приступа от

Гиперкалиемическая форма пароксизмальной миоплегии, или болезнь Гармстропа отличается слабостью мимической и
Гиперкалиемическая форма пароксизмальной миоплегии, или болезнь Гармстропа отличается слабостью мимической и

При гипокалиемическом параличе, обусловленном мутациями в гене CACNA1S (1q31-32),
дефектным оказывается
При гипокалиемическом параличе, обусловленном мутациями в гене CACNA1S (1q31-32), дефектным оказывается

Мутации в гене SCN4 (17q23.1), кодирующем альфа-4-субъединицу потенциал-зависимого натриевого канала, обнаруживаются
Мутации в гене SCN4 (17q23.1), кодирующем альфа-4-субъединицу потенциал-зависимого натриевого канала, обнаруживаются

В результате дефекта натриевого канала происходит усиление тока иона в клетку,
В результате дефекта натриевого канала происходит усиление тока иона в клетку,

Наследственные миастении
Наследственные миастении

В разделе наследственных нейромышечных заболеваний традиционно рассматривают миастению, или болезнь Эрба.
В разделе наследственных нейромышечных заболеваний традиционно рассматривают миастению, или болезнь Эрба.

Миастения клинически характеризуется нарастающей при выполнении движений мышечной слабостью, которая может
Миастения клинически характеризуется нарастающей при выполнении движений мышечной слабостью, которая может

Различают миастению генерализованную и локальную с поражением глазодвигательных нервов (глазная форма)
Различают миастению генерализованную и локальную с поражением глазодвигательных нервов (глазная форма)

Опасны миастенические кризы, когда внезапно развиваются генерализованная общая слабость и выраженные
Опасны миастенические кризы, когда внезапно развиваются генерализованная общая слабость и выраженные

Как и для многих других аутоиммунных заболеваний, генетическими факторами риска миастении
Как и для многих других аутоиммунных заболеваний, генетическими факторами риска миастении

В патогенезе аутоиммунной реакции основную роль играет тимус.
При патологии тимоцитов
В патогенезе аутоиммунной реакции основную роль играет тимус. При патологии тимоцитов

Патогенетическая терапия: антихолинэстеразные препараты (прозерин, оксазил, каллимин), курсы стероидной терапии.
При
Патогенетическая терапия: антихолинэстеразные препараты (прозерин, оксазил, каллимин), курсы стероидной терапии. При

Редкие врожденные миастенические синдромы – это моногенные формы миастении. Их классифицируют
Редкие врожденные миастенические синдромы – это моногенные формы миастении. Их классифицируют

Пресинаптический миастенически синдром, сопровождающийся эпизодической атаксией, обусловлен мутациями в гене
CHAT
Пресинаптический миастенически синдром, сопровождающийся эпизодической атаксией, обусловлен мутациями в гене CHAT

Постсинаптический синдром по характеру кинетического дефекта ацетилхолинового рецептора делится на два
Постсинаптический синдром по характеру кинетического дефекта ацетилхолинового рецептора делится на два

И в том, и в другом случае мутантными оказываются гены различных
И в том, и в другом случае мутантными оказываются гены различных

При этом причиной развития варианта с быстрым потоком являются инактивирующие мутации,
При этом причиной развития варианта с быстрым потоком являются инактивирующие мутации,

При варианте с медленным потоком мутации в генах различных субъединиц ацетилхолинового
При варианте с медленным потоком мутации в генах различных субъединиц ацетилхолинового

В некоторых случаях у больных с врожденным миастеническим синдромом обнаруживают мутации
В некоторых случаях у больных с врожденным миастеническим синдромом обнаруживают мутации

Синаптический вариант врожденного миастенического синдрома обусловлен недостаточностью синаптической ацетилхолинэстеразы, причиной развития
Синаптический вариант врожденного миастенического синдрома обусловлен недостаточностью синаптической ацетилхолинэстеразы, причиной развития

При нарушении структуры коллагена Q не происходит встраивания ацетилхолинэстеразы в базальную
При нарушении структуры коллагена Q не происходит встраивания ацетилхолинэстеразы в базальную
 Профессия - диетолог
Профессия - диетолог Гигиенические требования к планировке оборудования и содержания детских и подростковых учреждений (5)
Гигиенические требования к планировке оборудования и содержания детских и подростковых учреждений (5) Принципы рациональной антибиотикотерапии
Принципы рациональной антибиотикотерапии Отделяемое мочеполовых органов
Отделяемое мочеполовых органов Все виды шрамов. Можно ли избавиться от шрамов и как
Все виды шрамов. Можно ли избавиться от шрамов и как Балаларда темір жеткіліксіздік анемиясы
Балаларда темір жеткіліксіздік анемиясы Патофизиология эндокринной системы
Патофизиология эндокринной системы СНІД – загроза людству
СНІД – загроза людству Злоякісні лімфоми. Лімфогранульоматоз, неходжкінські лімфоми
Злоякісні лімфоми. Лімфогранульоматоз, неходжкінські лімфоми Механизм действия, показания и противопоказания к применению антикоагулянтов, методы контроля; побочные эффекты
Механизм действия, показания и противопоказания к применению антикоагулянтов, методы контроля; побочные эффекты Эпидемиологическая ситуация по ВИЧ - инфекции на территории города Пыть-Яха КУ. Центр СПИД. Филиал в г. Пыть-Яхе
Эпидемиологическая ситуация по ВИЧ - инфекции на территории города Пыть-Яха КУ. Центр СПИД. Филиал в г. Пыть-Яхе Неонатальный скрининг ВПС
Неонатальный скрининг ВПС Деструктивные формы туберкулеза
Деструктивные формы туберкулеза Лекарственная безопасность
Лекарственная безопасность Печень. Участие печени в процессах гомеостаза/гомеокинеза организма
Печень. Участие печени в процессах гомеостаза/гомеокинеза организма Гломерулонефрит. Клиническая классификация. Диагностика. Принципы лечения
Гломерулонефрит. Клиническая классификация. Диагностика. Принципы лечения Ұзақ өмір сүрушілік, қартаю.Клиникалық және этикалық аспектілері. Ұзақ өмір сүрушілік феноменінің генетикалық механизмдері
Ұзақ өмір сүрушілік, қартаю.Клиникалық және этикалық аспектілері. Ұзақ өмір сүрушілік феноменінің генетикалық механизмдері Денсаулық сақтауды қаржыландыру
Денсаулық сақтауды қаржыландыру Пожари. Оказана помощ
Пожари. Оказана помощ Тейлериидозы животных (тейлериоз и нутталлиоз)
Тейлериидозы животных (тейлериоз и нутталлиоз) Терминальные состояния
Терминальные состояния Пациенты с заболеванием органов дыхания: особенности ухода
Пациенты с заболеванием органов дыхания: особенности ухода Эффективность остеопатической коррекции при перинатальной энцефалопатии у недоношенных детей
Эффективность остеопатической коррекции при перинатальной энцефалопатии у недоношенных детей Увеличение щитовидной железы
Увеличение щитовидной железы Нейтрофильные гранулоциты. Нейтропении. Тяжелая врожденная нейтропения
Нейтрофильные гранулоциты. Нейтропении. Тяжелая врожденная нейтропения Методы нейровизуализации гидроцефалии и микроцефалии
Методы нейровизуализации гидроцефалии и микроцефалии Оппортунистические заболевания при ВИЧ‐инфекции
Оппортунистические заболевания при ВИЧ‐инфекции Возможности деятельности Федерации по оптимизации использования ресурсов при обеспечении экстренной медицинской помощи
Возможности деятельности Федерации по оптимизации использования ресурсов при обеспечении экстренной медицинской помощи