- Наследственные онкологические синдромы. Наследственные опухоли. Семинар 10

Содержание
- 3. Вклад генетических и средовых факторов при развитии спорадических опухолей По данным регистров близнецов Швеции, Дании и
- 4. Механизм развития спорадических и наследственных опухолей Спорадические Наследственные
- 5. Наследственные опухолевые синдромы – группа заболеваний, проявление которых заключается в передаче из поколения в поколение предрасположенности
- 7. Рак молочной железы (РМЖ) и рак яичников (РЯ) представляют собой важную социально-медицинскую проблему в связи с
- 8. Признаки наследственной опухоли молочной железы и яичников несколько случаев опухолей молочной железы и яичника у кровных
- 9. Гены BRCA1 и BRCA2 (BREAST CANCER GENES 1&2) BRCA1 и BRCA2 кодируют аминокислотные последовательности ядерных белков,
- 13. Мутации в генах BRCA1 и BRCA2
- 14. Ген CHEK2 (CHECKPOINT KINASE 2) Ген CHEK2 кодирует белок чекпойнт-киназу 2. Продукт гена CHEK2 участвует в
- 15. Ген NBS1 (NIJMEGEN BREAKAGE SYNDROME) Ген NBS1 кодирует белок нибрин, который участвует в регуляции клеточного цикла,
- 16. 10 наиболее частых для российской популяции мутаций в генах BRCA1, BRCA2, СНЕК2 и NBS1 ∙ BRCA1
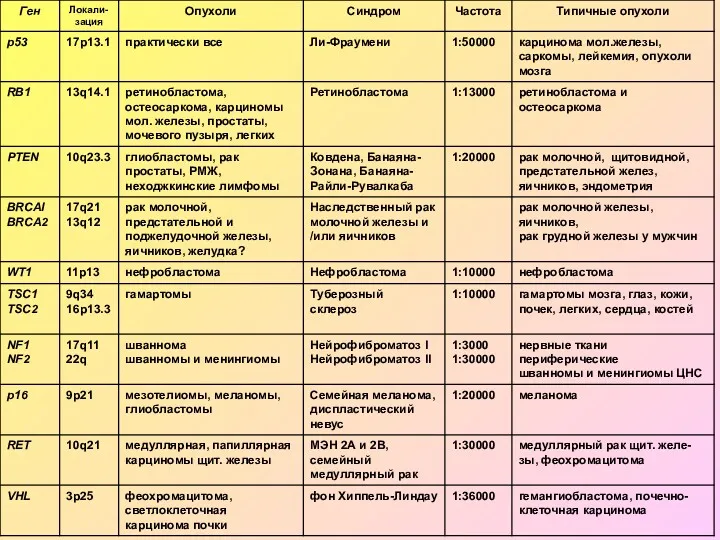
- 17. Синдромы наследственного рака молочной железы
- 18. Увеличивается количество генов, которые вызывают уже известные наследственные синдромы с опухолями определенной локализации Опухоли другой локализации
- 19. Исследовать все известные гены, имеющие отношение к развитию наследственного рака желудка Исследовать всю кодирующую последовательность генов,
- 23. МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ АСПЕКТЫ НАСЛЕДСТВЕННОГО РАКА ТОЛСТОЙ КИШКИ
- 24. рак толстой кишки 2009 г. В России: зарегистрировано 57 363 случаев РТК Умерло: 38 343 больных
- 25. Наследственный РТК Наследственный Неполипозный Рак Толстой Кишки – ННПРТК, синдром Линча Семейный аденоматозный полипоз толстой кишки
- 26. Наследственный неполипозный рак толстой кишки (ННПРТК) История открытия ННПРТК связана с именем американского ученого Генри Линча,
- 27. ННПРТК характеризуется преимущественно поражением правых отделов ободочной кишки На долю РТК при синдроме Линча приходится 1-3%
- 28. Амстердамские критерии I (1991 г. ): Молодой возраст возникновения заболевания (до 50 лет) Наличие 3 или
- 29. Критерии Bethesda (2004г.) 1. Колоректальный рак в возрасте до 50 лет 2. Наличие синхронных, метахронных опухолей
- 31. Эффективность критериев У пациентов соответствующих Амстердамским критериям частота обнаружения наследственных мутаций около 50% У пациентов соответствующих
- 32. Генетика синдрома Линча Причина возникновения – наследственная мутация в одном из генов системы репарации неправильно спаренных
- 33. Частоты мутаций в генах MMR при наследственном неполипозном раке толстой кишки – InSiGHT database MLH1 –
- 34. К злокачественной трансформации клеток приводят накопление критического числа мутаций в отсутствие адекватной системы репарации ДНК Система
- 35. Модель репарации неспаренных оснований
- 36. Механизм микросателлитной нестабильности Молекулярной характеристикой опухолей ННПРТК является проявление микросателлитной нестабильности
- 37. Экзонная структура генов MLH1 и MSH2 Так как частых мутаций из более чем 1000 известных к
- 38. Молекулярно-генетическая диагностика ННПРТК 1 этап: Исследование микросателлитной нестабильности в опухоли – MSI: MSI-S (stable) или MSI-H
- 39. РИСК РАЗВИТИЯ ОПУХОЛЕЙ РАЗНОЙ ЛОКАЛИЗАЦИИ У НОСИТЕЛЕЙ МУТАЦИИ В ГЕНАХ MMR
- 40. Колоноскопия у здоровых носителей мутации (с 20-25 лет до 80 лет, 1 раз в 2-3 года)
- 41. Критерии отбора пациентов с ННПРТК для генетического тестирования Для всех пациентов со спорадическим РТК в возрасте
- 42. Семеный аденоматоз толстой кишки САТК Тяжелое наследственное заболевание, характеризуется - множественными аденоматозными полипами толстой кишки и
- 43. Классическая и тяжелая формы полипоза Развитие сотен и даже тысяч полипов Рост полипов может начинаться уже
- 44. Ослабленная форма полипоза Наличие менее 100 аденоматозных полипов Более поздние сроки возникновения и озлокачествления полипов Недостаточное
- 45. Структура гена APC (5q21) Ген APC содержит 15 кодирующих экзонов (8535 нуклеотидных пар) и кодирует белок,
- 46. Функционирование гена АРС в Wnt-пути Опухолевые клетки толстой кишки, имеющие мутацию гена АРС, имеют высокий уровень
- 47. Типы патогенных мутаций в гене АРС Sarah E. Kerr APC Germline Mutations in Individuals Being Evaluated
- 48. Развитие опухолей в зависимости от локализации мутации
- 49. Зависимость клинической картины от локализации мутаций в гене APC
- 50. Ген MYH Расположен на 1 хромосоме Ген включает 16 кодирующих экзонов Продукт гена MYH участвует в
- 51. у пациентов с классической или тяжелой формой заболевания – исследование гена APC У больных с ослабленной
- 52. Редкие синдромы с РТК Синдром Пейтца-Егерса (PJS) STK11 Синдром Коудена PTEN Семейный ювенильный SMAD4 полипоз (FJP)
- 53. Молекулярный генез колоректального рака спорадический наследственный Семейный аденоматозный полипоз APC Наследственный неполипозный колоректальный рак MLH1 Эпигенетические
- 54. Соматические мутации в генах В большинстве опухолей мутированы гены APC и TP53 KRAS – 35-45% BRAF–
- 55. MSI-H опухоли не склонны к метастазированию, имеют благоприятный прогноз наличие мутации в гене KRAS сопровождается резистентностью
- 56. Предиктивное и прогностическое значение мутационного статуса опухоли Ген KRAS мутации в 12,13,61 кодонах– 35-45% Ретроспективный анализ
- 57. МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ АСПЕКТЫ НАСЛЕДСТВЕННОГО РАКА ЖЕЛУДКА
- 58. Наиболее известной жертвой семейного РЖ является французский император Наполеон Бонапарт, скончавшийся именно от этого заболевания, и
- 59. Наследственный диффузный рак желудка Семейное обогащение наблюдается у 15% больных РЖ, но только около 5% случаев
- 60. В 2004 году были предложены критерии для характеристики больных и их семей, которые связаны с повышенной
- 61. Кадгерин-катениновые взаимодействия необходимы для образования и поддержания межклеточных контактов
- 62. Ген CDH1 локализуется на хромосоме 16q22.1, занимая объем около 100 кб. CDH1 содержит 16 экзонов и
- 63. Наследственные формы рака желудка
- 66. Спорадический рак инициируется накоплением молекулярных повреждений в геноме соматических клеток определенных тканей и органов в процессе
- 71. Скачать презентацию

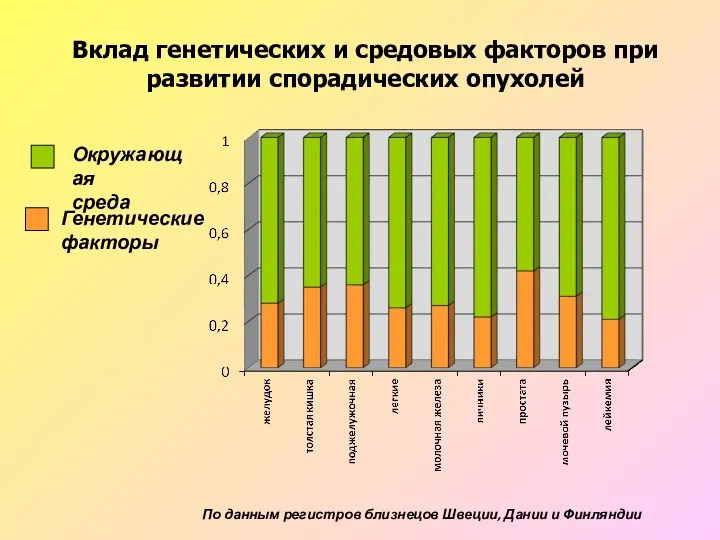
Вклад генетических и средовых факторов при развитии спорадических опухолей
По данным регистров
Вклад генетических и средовых факторов при развитии спорадических опухолей
По данным регистров
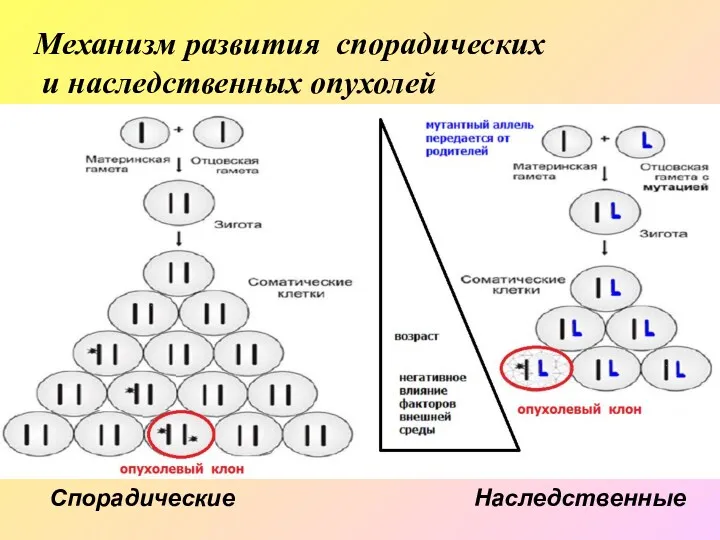
Механизм развития спорадических
и наследственных опухолей
Спорадические Наследственные
Механизм развития спорадических
и наследственных опухолей
Спорадические Наследственные

Наследственные опухолевые синдромы – группа заболеваний, проявление которых заключается в передаче
Наследственные опухолевые синдромы – группа заболеваний, проявление которых заключается в передаче

Рак молочной железы (РМЖ) и рак яичников (РЯ) представляют собой важную
Рак молочной железы (РМЖ) и рак яичников (РЯ) представляют собой важную

Признаки наследственной опухоли молочной железы и яичников
несколько случаев опухолей молочной железы
Признаки наследственной опухоли молочной железы и яичников
несколько случаев опухолей молочной железы

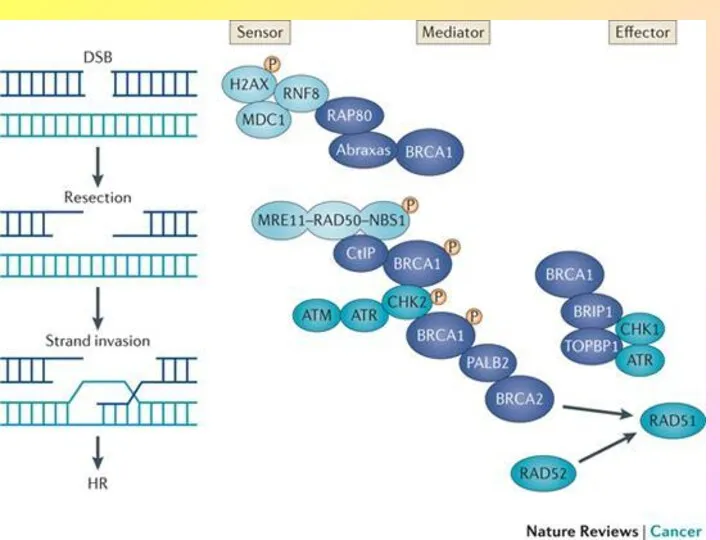
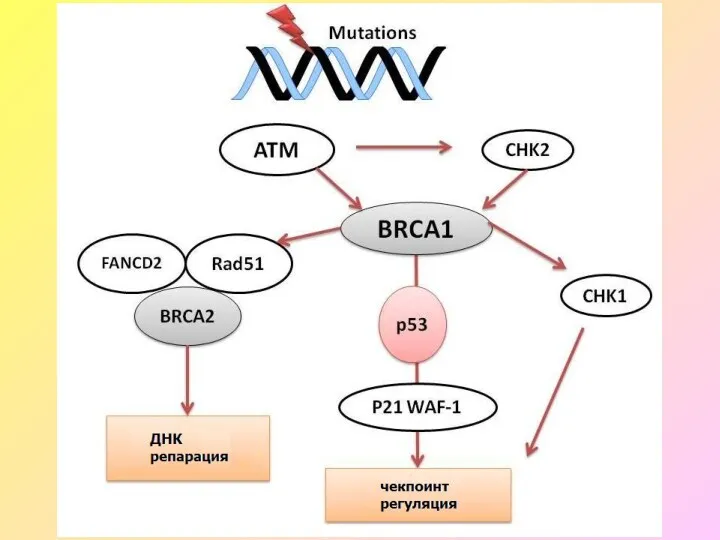
Гены BRCA1 и BRCA2 (BREAST CANCER GENES 1&2)
BRCA1 и BRCA2 кодируют аминокислотные последовательности ядерных белков, которые участвуют
Гены BRCA1 и BRCA2 (BREAST CANCER GENES 1&2)
BRCA1 и BRCA2 кодируют аминокислотные последовательности ядерных белков, которые участвуют
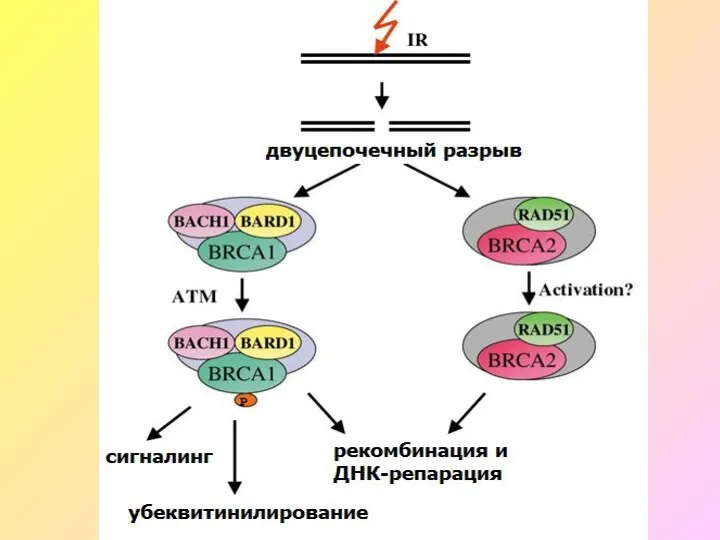
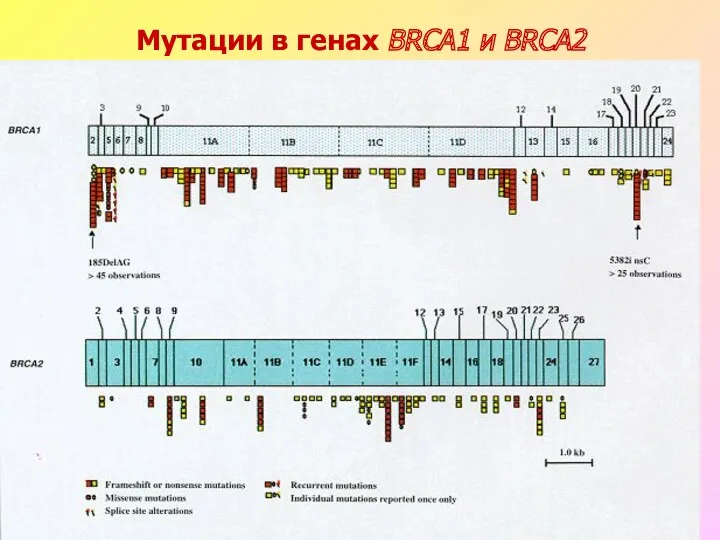
Мутации в генах BRCA1 и BRCA2
Мутации в генах BRCA1 и BRCA2

Ген CHEK2 (CHECKPOINT KINASE 2)
Ген CHEK2 кодирует белок чекпойнт-киназу 2. Продукт гена CHEK2
Ген CHEK2 (CHECKPOINT KINASE 2)
Ген CHEK2 кодирует белок чекпойнт-киназу 2. Продукт гена CHEK2

Ген NBS1 (NIJMEGEN BREAKAGE SYNDROME)
Ген NBS1 кодирует белок нибрин, который участвует в регуляции
Ген NBS1 (NIJMEGEN BREAKAGE SYNDROME)
Ген NBS1 кодирует белок нибрин, который участвует в регуляции

10 наиболее частых для российской популяции мутаций в генах BRCA1, BRCA2,
10 наиболее частых для российской популяции мутаций в генах BRCA1, BRCA2,

Синдромы наследственного рака молочной железы
Синдромы наследственного рака молочной железы
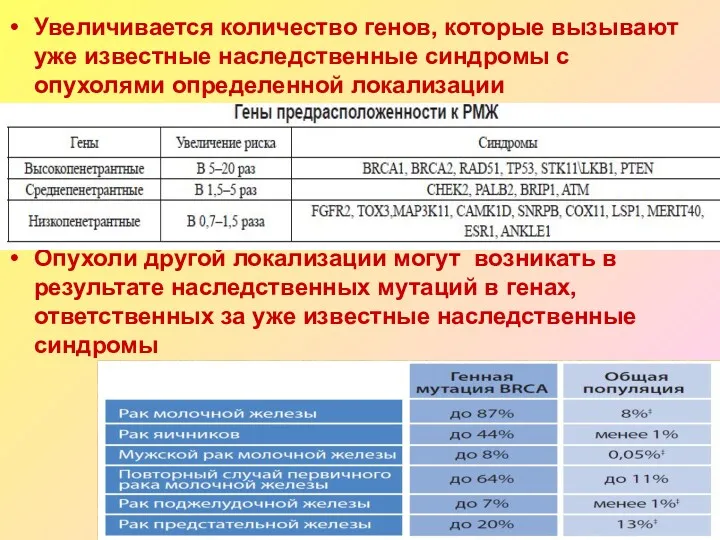
Увеличивается количество генов, которые вызывают уже известные наследственные синдромы с опухолями
Увеличивается количество генов, которые вызывают уже известные наследственные синдромы с опухолями
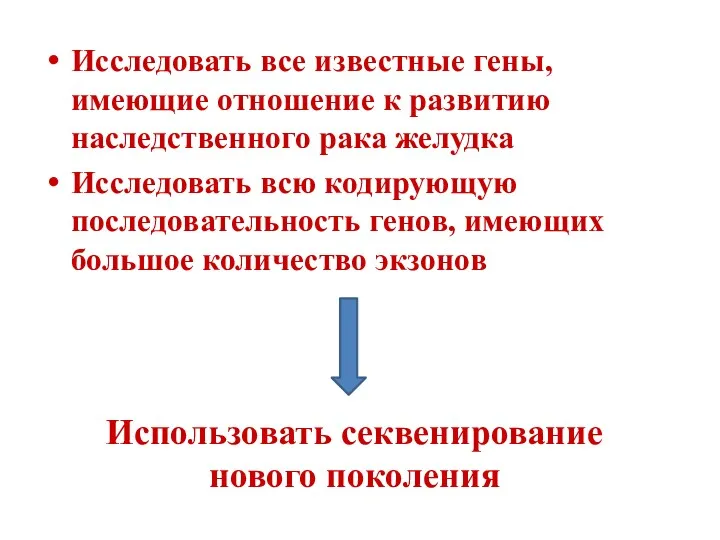
Исследовать все известные гены, имеющие отношение к развитию наследственного рака желудка
Исследовать
Исследовать все известные гены, имеющие отношение к развитию наследственного рака желудка
Исследовать
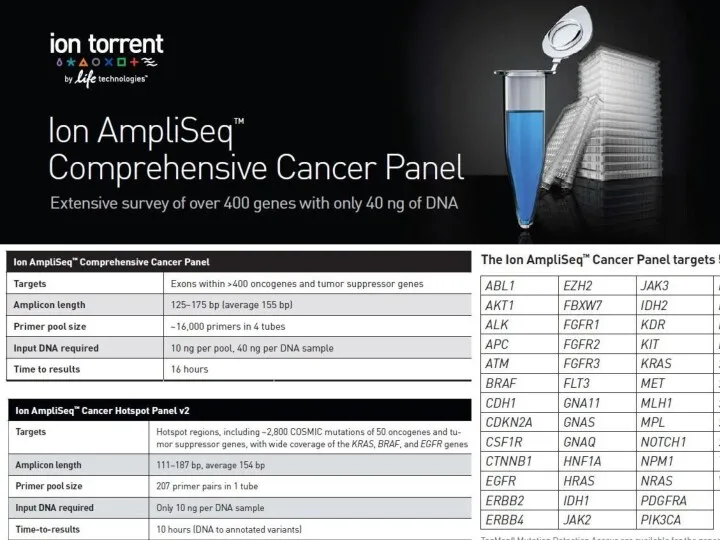
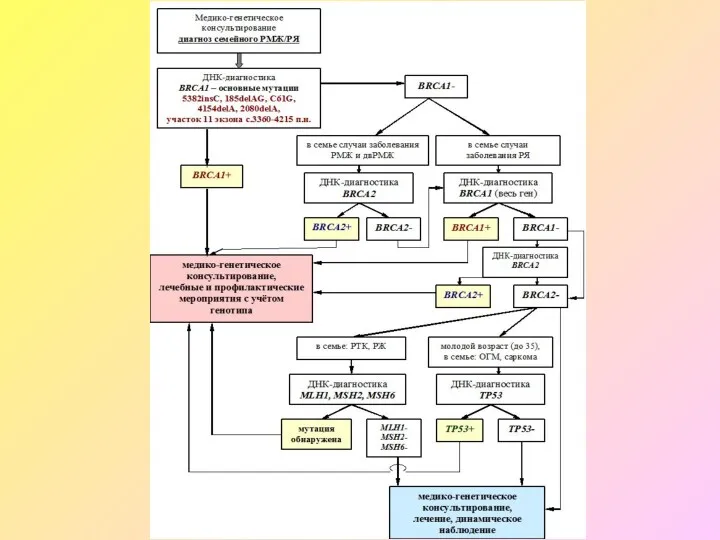


МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ АСПЕКТЫ НАСЛЕДСТВЕННОГО РАКА ТОЛСТОЙ КИШКИ
МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ АСПЕКТЫ НАСЛЕДСТВЕННОГО РАКА ТОЛСТОЙ КИШКИ

рак толстой кишки
2009 г. В России:
зарегистрировано 57 363 случаев РТК
Умерло:
рак толстой кишки
2009 г. В России:
зарегистрировано 57 363 случаев РТК
Умерло:
Наследственный РТК
Наследственный Неполипозный Рак Толстой Кишки –
ННПРТК, синдром Линча
Семейный аденоматозный
Наследственный РТК
Наследственный Неполипозный Рак Толстой Кишки –
ННПРТК, синдром Линча
Семейный аденоматозный

Наследственный неполипозный рак толстой кишки (ННПРТК)
История открытия ННПРТК связана с
Наследственный неполипозный рак толстой кишки (ННПРТК)
История открытия ННПРТК связана с
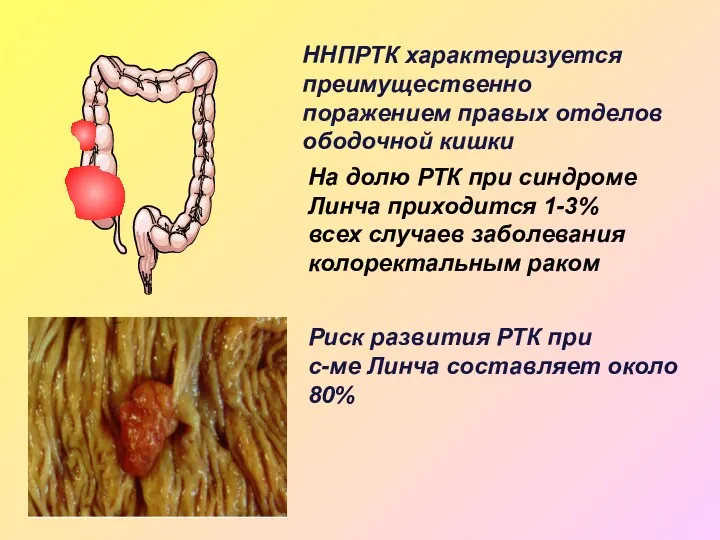
ННПРТК характеризуется
преимущественно поражением правых отделов ободочной кишки
На долю РТК при синдроме
ННПРТК характеризуется
преимущественно поражением правых отделов ободочной кишки
На долю РТК при синдроме

Амстердамские критерии I (1991 г. ):
Молодой возраст возникновения заболевания (до
Амстердамские критерии I (1991 г. ):
Молодой возраст возникновения заболевания (до

Критерии Bethesda (2004г.)
1. Колоректальный рак в возрасте до 50 лет
2. Наличие
Критерии Bethesda (2004г.)
1. Колоректальный рак в возрасте до 50 лет
2. Наличие
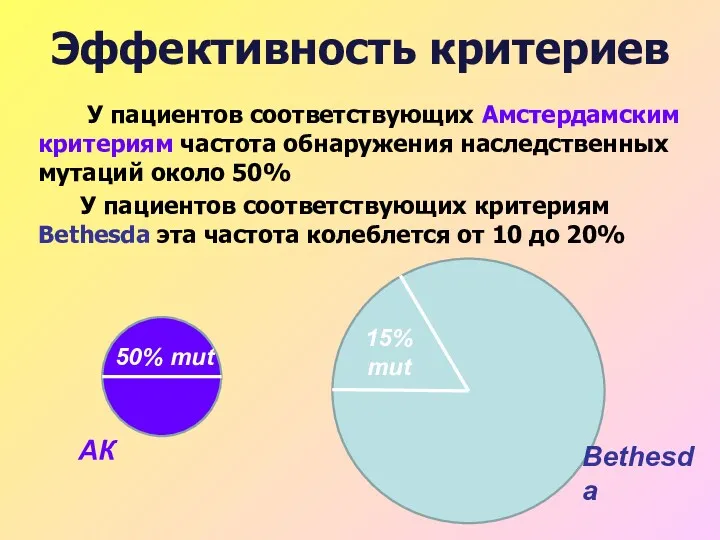
Эффективность критериев
У пациентов соответствующих Амстердамским критериям частота обнаружения наследственных мутаций
Эффективность критериев
У пациентов соответствующих Амстердамским критериям частота обнаружения наследственных мутаций

Генетика синдрома Линча
Причина возникновения – наследственная мутация в одном из генов
Генетика синдрома Линча
Причина возникновения – наследственная мутация в одном из генов

Частоты мутаций в генах MMR при наследственном неполипозном раке толстой кишки
Частоты мутаций в генах MMR при наследственном неполипозном раке толстой кишки
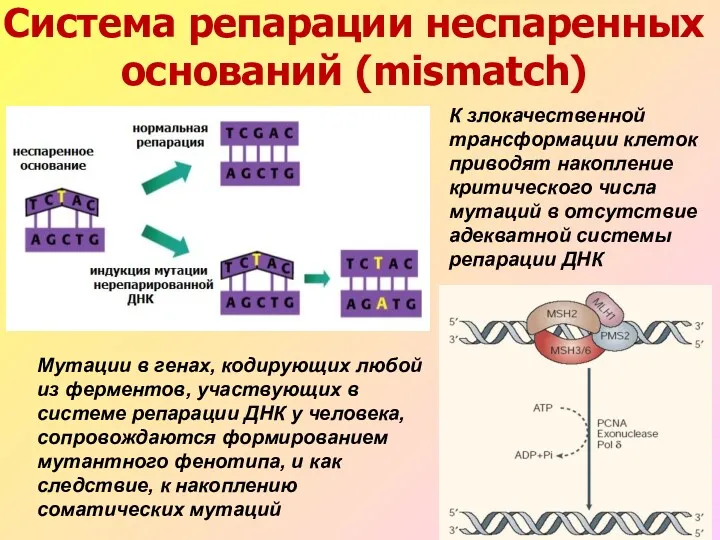
К злокачественной трансформации клеток приводят накопление критического числа мутаций в отсутствие
К злокачественной трансформации клеток приводят накопление критического числа мутаций в отсутствие
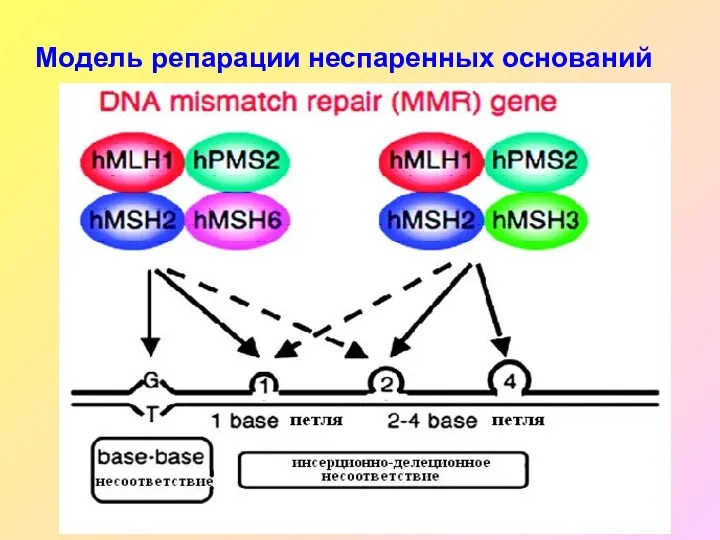
Модель репарации неспаренных оснований
Модель репарации неспаренных оснований
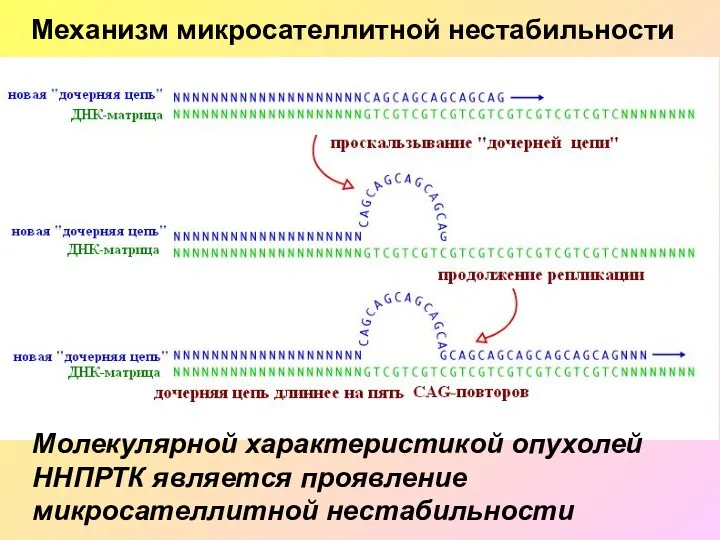
Механизм микросателлитной нестабильности
Молекулярной характеристикой опухолей ННПРТК является проявление микросателлитной нестабильности
Механизм микросателлитной нестабильности
Молекулярной характеристикой опухолей ННПРТК является проявление микросателлитной нестабильности
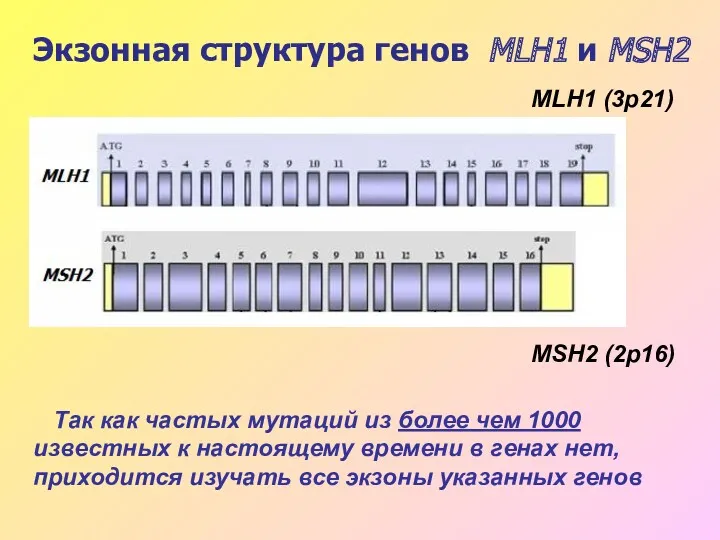
Экзонная структура генов MLH1 и MSH2
Так как частых мутаций из
Экзонная структура генов MLH1 и MSH2
Так как частых мутаций из

Молекулярно-генетическая диагностика ННПРТК
1 этап: Исследование микросателлитной нестабильности в опухоли – MSI:
Молекулярно-генетическая диагностика ННПРТК
1 этап: Исследование микросателлитной нестабильности в опухоли – MSI:
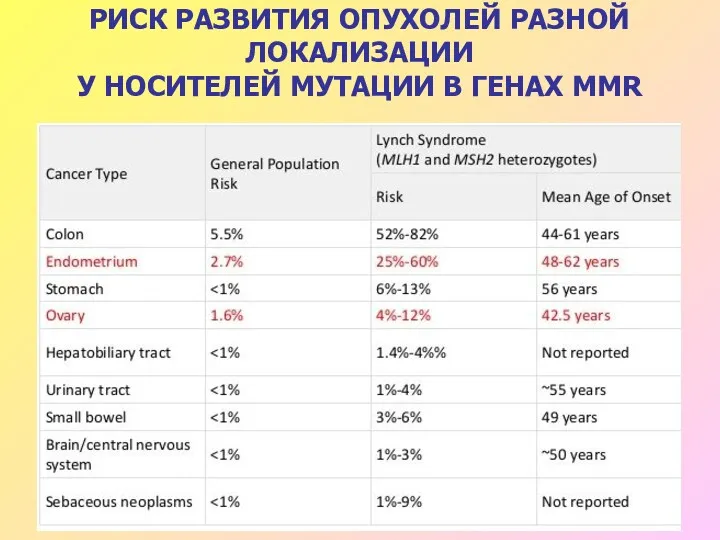
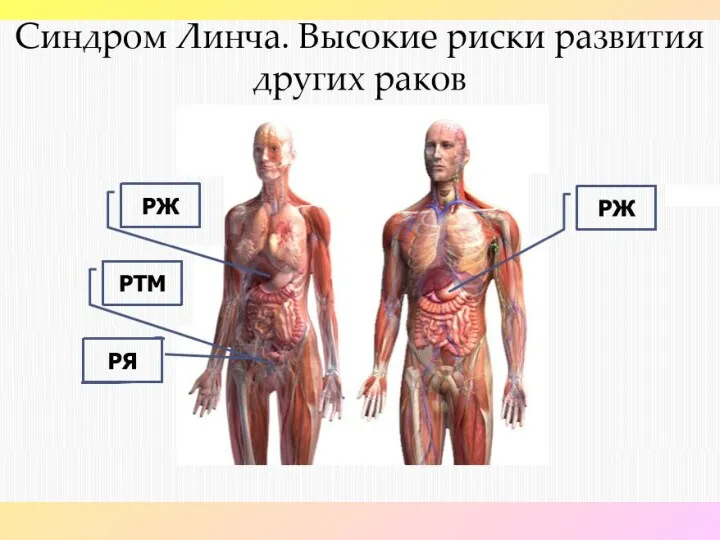
РИСК РАЗВИТИЯ ОПУХОЛЕЙ РАЗНОЙ ЛОКАЛИЗАЦИИ
У НОСИТЕЛЕЙ МУТАЦИИ В ГЕНАХ MMR
РИСК РАЗВИТИЯ ОПУХОЛЕЙ РАЗНОЙ ЛОКАЛИЗАЦИИ
У НОСИТЕЛЕЙ МУТАЦИИ В ГЕНАХ MMR

Колоноскопия у здоровых носителей мутации (с 20-25 лет до 80 лет,
Колоноскопия у здоровых носителей мутации (с 20-25 лет до 80 лет,

Критерии отбора пациентов
с ННПРТК для генетического тестирования
Для всех пациентов
Критерии отбора пациентов
с ННПРТК для генетического тестирования
Для всех пациентов

Семеный аденоматоз толстой кишки
САТК
Тяжелое наследственное заболевание, характеризуется - множественными аденоматозными
Семеный аденоматоз толстой кишки
САТК
Тяжелое наследственное заболевание, характеризуется - множественными аденоматозными

Классическая и тяжелая формы полипоза
Развитие сотен и даже тысяч полипов
Рост полипов
Классическая и тяжелая формы полипоза
Развитие сотен и даже тысяч полипов
Рост полипов
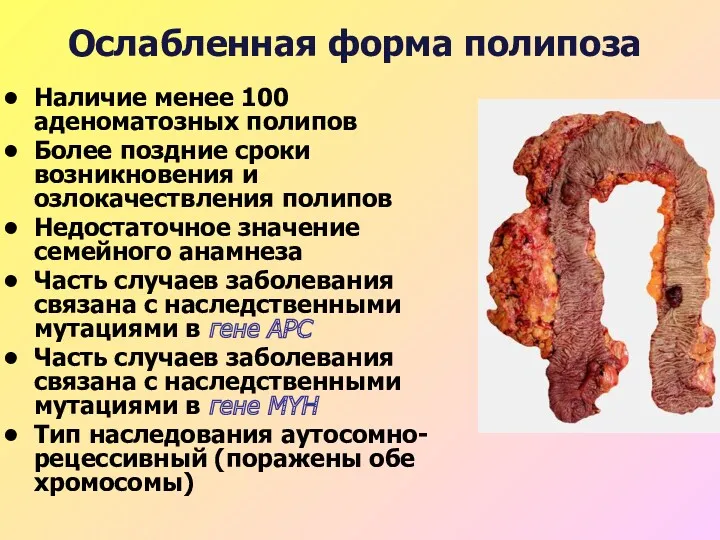
Ослабленная форма полипоза
Наличие менее 100 аденоматозных полипов
Более поздние сроки возникновения и
Ослабленная форма полипоза
Наличие менее 100 аденоматозных полипов
Более поздние сроки возникновения и

Структура гена APC (5q21)
Ген APC содержит 15 кодирующих экзонов
Структура гена APC (5q21)
Ген APC содержит 15 кодирующих экзонов
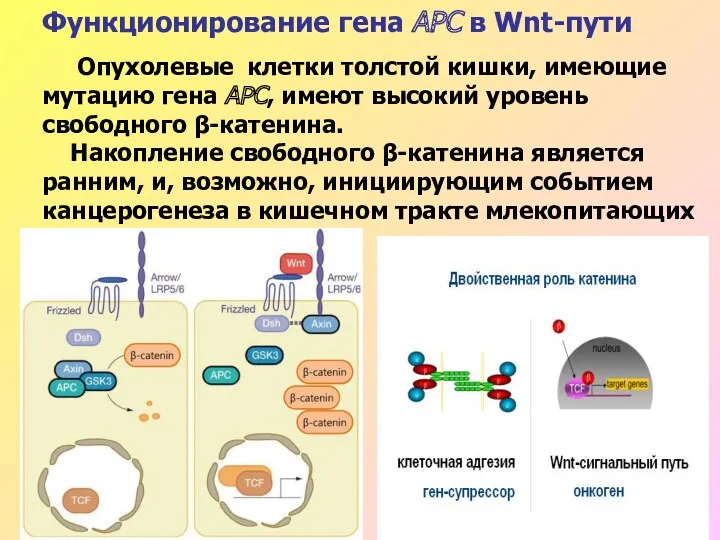
Функционирование гена АРС в Wnt-пути
Опухолевые клетки толстой кишки, имеющие мутацию
Функционирование гена АРС в Wnt-пути
Опухолевые клетки толстой кишки, имеющие мутацию

Типы патогенных мутаций в гене АРС
Sarah E. Kerr
APC Germline
Типы патогенных мутаций в гене АРС
Sarah E. Kerr
APC Germline
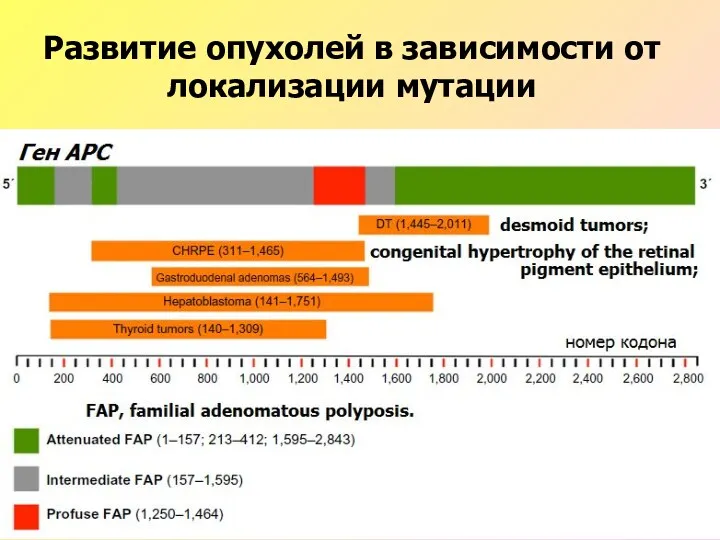
Развитие опухолей в зависимости от локализации мутации
Развитие опухолей в зависимости от локализации мутации

Зависимость клинической картины от локализации мутаций в гене APC
Зависимость клинической картины от локализации мутаций в гене APC

Ген MYH
Расположен на 1 хромосоме
Ген включает 16 кодирующих экзонов
Продукт гена
Ген MYH
Расположен на 1 хромосоме
Ген включает 16 кодирующих экзонов
Продукт гена

у пациентов с классической или тяжелой формой заболевания – исследование гена

Редкие синдромы с РТК
Синдром Пейтца-Егерса (PJS) STK11
Синдром Коудена PTEN
Семейный ювенильный SMAD4
полипоз
Редкие синдромы с РТК
Синдром Пейтца-Егерса (PJS) STK11
Синдром Коудена PTEN
Семейный ювенильный SMAD4
полипоз
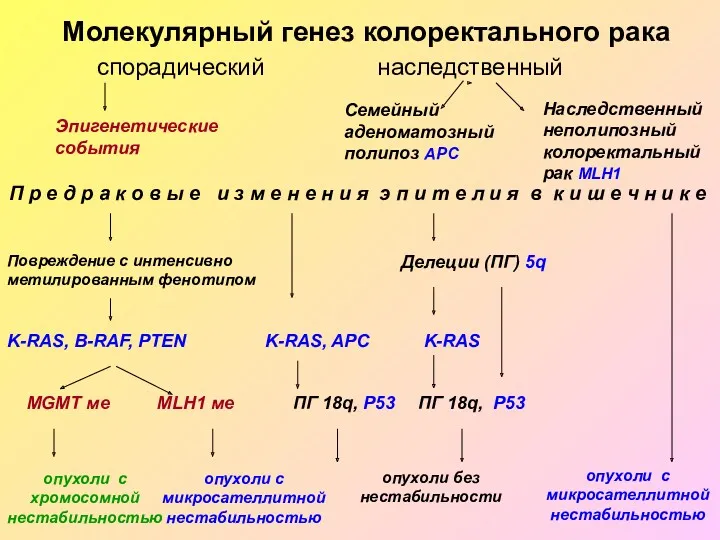
Молекулярный генез колоректального рака
спорадический
наследственный
Семейный аденоматозный полипоз APC
Наследственный неполипозный колоректальный
Молекулярный генез колоректального рака
спорадический
наследственный
Семейный аденоматозный полипоз APC
Наследственный неполипозный колоректальный

Соматические мутации в генах
В большинстве опухолей мутированы гены APC и
Соматические мутации в генах
В большинстве опухолей мутированы гены APC и

MSI-H опухоли не склонны к метастазированию, имеют благоприятный прогноз
наличие
MSI-H опухоли не склонны к метастазированию, имеют благоприятный прогноз
наличие

Предиктивное и прогностическое значение мутационного статуса опухоли
Ген KRAS
мутации в 12,13,61 кодонах–
Предиктивное и прогностическое значение мутационного статуса опухоли
Ген KRAS
мутации в 12,13,61 кодонах–

МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ АСПЕКТЫ НАСЛЕДСТВЕННОГО РАКА ЖЕЛУДКА
МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ АСПЕКТЫ НАСЛЕДСТВЕННОГО РАКА ЖЕЛУДКА

Наиболее известной жертвой семейного РЖ является французский император Наполеон Бонапарт, скончавшийся
Наиболее известной жертвой семейного РЖ является французский император Наполеон Бонапарт, скончавшийся

Наследственный диффузный рак желудка
Семейное обогащение наблюдается у 15% больных РЖ, но
Наследственный диффузный рак желудка
Семейное обогащение наблюдается у 15% больных РЖ, но

В 2004 году были предложены критерии для характеристики больных и их
В 2004 году были предложены критерии для характеристики больных и их
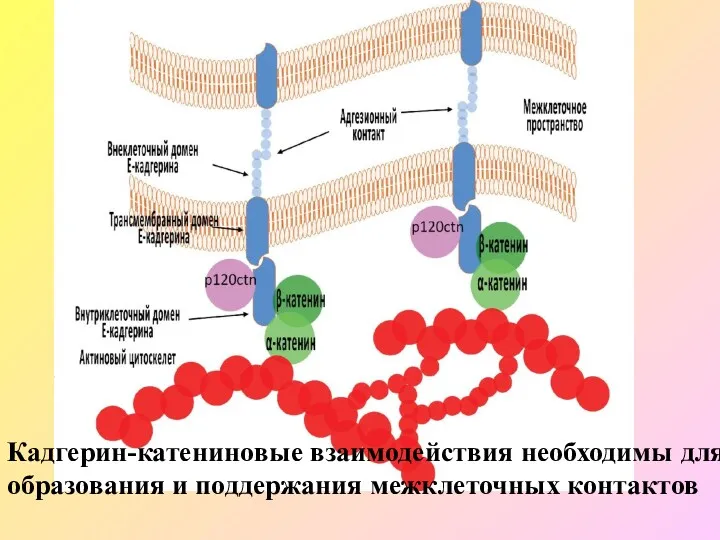
Кадгерин-катениновые взаимодействия необходимы для образования и поддержания межклеточных контактов
Кадгерин-катениновые взаимодействия необходимы для образования и поддержания межклеточных контактов

Ген CDH1 локализуется на хромосоме 16q22.1, занимая объем около 100 кб.
Ген CDH1 локализуется на хромосоме 16q22.1, занимая объем около 100 кб.
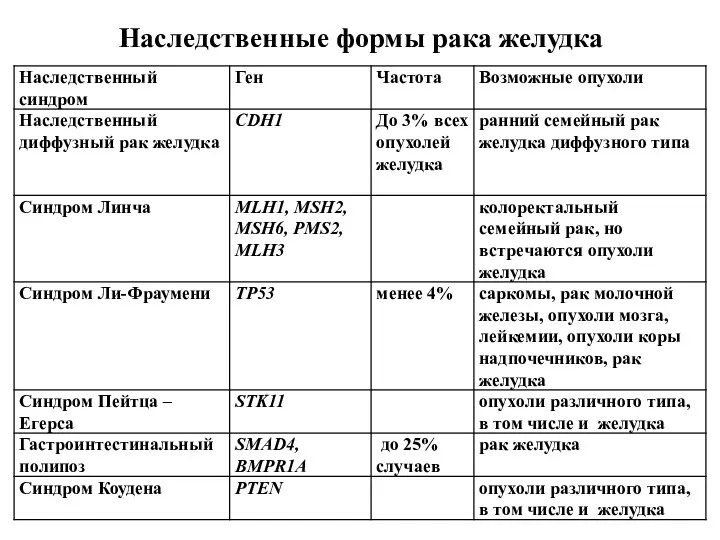
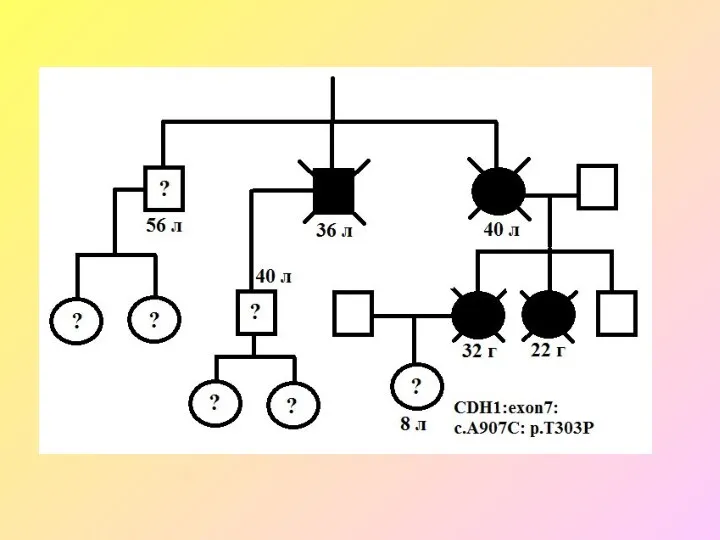
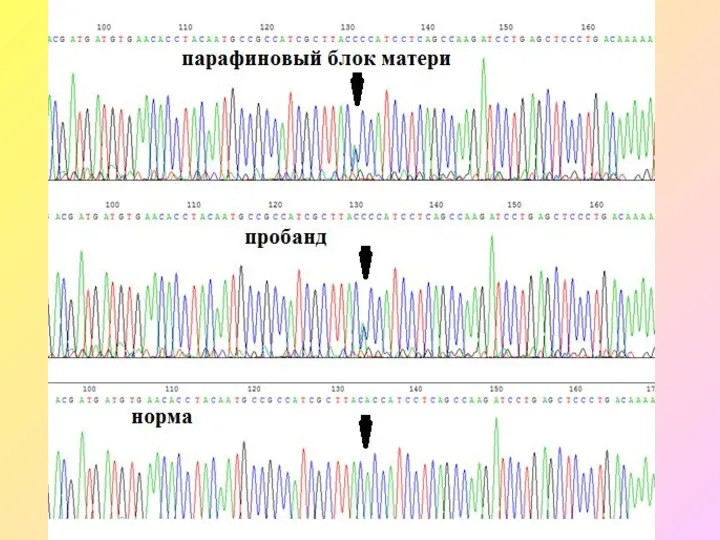
Наследственные формы рака желудка
Наследственные формы рака желудка
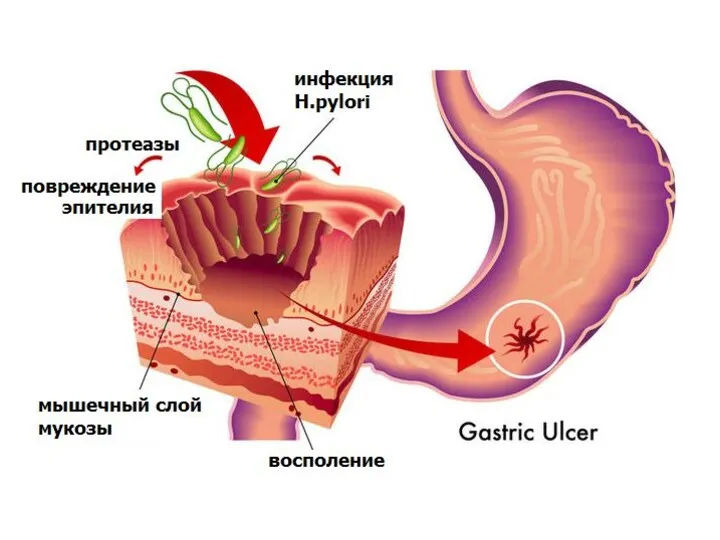
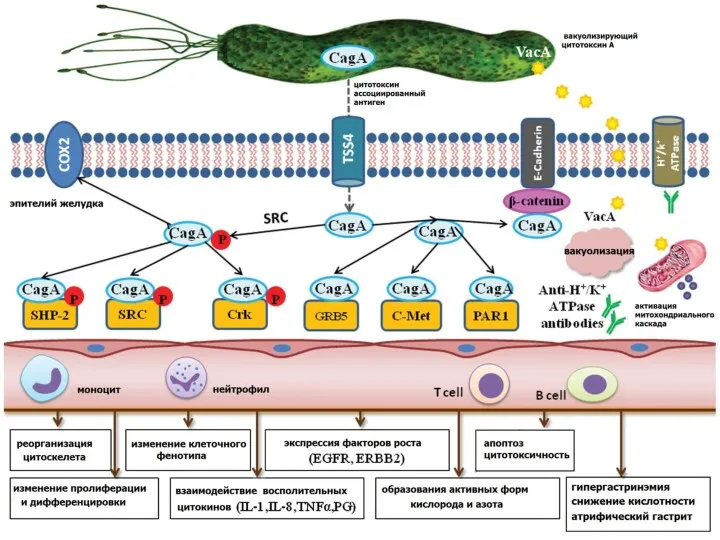
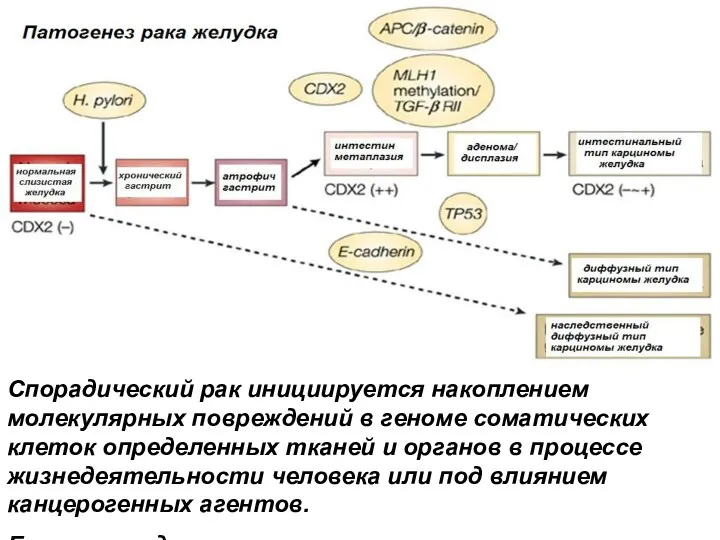
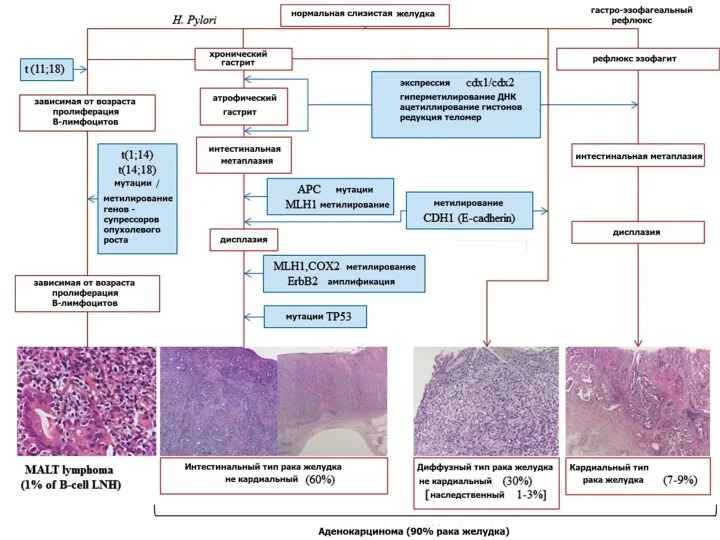
Спорадический рак инициируется накоплением молекулярных повреждений в геноме соматических клеток определенных
Спорадический рак инициируется накоплением молекулярных повреждений в геноме соматических клеток определенных
 Healthy lifestyle and personal hygiene. Psychohygiene. Physical culture and bases of tempering
Healthy lifestyle and personal hygiene. Psychohygiene. Physical culture and bases of tempering Методика адаптивного физического воспитания для обучающихся с задержкой психического развития
Методика адаптивного физического воспитания для обучающихся с задержкой психического развития Отит. Виды отита
Отит. Виды отита Если хочешь быть здоров
Если хочешь быть здоров Стабилизаторы мембран тучных клеток
Стабилизаторы мембран тучных клеток Медицинская реабилитация в онкологиии
Медицинская реабилитация в онкологиии Бүйрек және зәр шығару жолдарының ауыруы бар науқастардың күтімі және бақылауы
Бүйрек және зәр шығару жолдарының ауыруы бар науқастардың күтімі және бақылауы Меланома кожи
Меланома кожи Иммунология
Иммунология Боковой амиотрофический склероз
Боковой амиотрофический склероз Врожденный сифилис
Врожденный сифилис Гистофизиология покровных и железистых эпителиев
Гистофизиология покровных и железистых эпителиев Физиология сосудов
Физиология сосудов Общие основы ЛФК
Общие основы ЛФК Проблемные вопросы качественной организации работы лаборатории
Проблемные вопросы качественной организации работы лаборатории Неінфекційні захворювання. Урок №12. Здоров'я, безпека та добробут. 6 клас
Неінфекційні захворювання. Урок №12. Здоров'я, безпека та добробут. 6 клас Di̇yabet & astracheck pro kan şekeri̇ ölçüm si̇stemleri̇
Di̇yabet & astracheck pro kan şekeri̇ ölçüm si̇stemleri̇ Созылмалы холецистит. Постхолецистоэктомия
Созылмалы холецистит. Постхолецистоэктомия Covid-19. Симптомы и признаки коронавируса у человека
Covid-19. Симптомы и признаки коронавируса у человека Основы радиационной гигиены
Основы радиационной гигиены Фармакокинетика
Фармакокинетика Одонтопрепарирование при ортопедическом лечении различными конструкциями зубных протезов
Одонтопрепарирование при ортопедическом лечении различными конструкциями зубных протезов Психологические особенности личности инсультного больного
Психологические особенности личности инсультного больного Гинекологическая патология
Гинекологическая патология Қозғалыс. Ерікті қимыл-қозғалыс. Орталық және шеткі паралич
Қозғалыс. Ерікті қимыл-қозғалыс. Орталық және шеткі паралич Проба Манту
Проба Манту Геморрой — царская болезнь
Геморрой — царская болезнь Лечебная физкультура при гинекологических заболеваниях
Лечебная физкультура при гинекологических заболеваниях