- Нейроофтальмологические нарушения при нейромышечных заболеваниях

Содержание
- 2. Нервно-мышечные заболевания – обобщённое название различных видов первичной и вторичной мышечной патологии , которые включают: Наследственные,
- 3. К прогрессирующим мышечным дистрофиям относятся: Первичные миопатии(амиотрофии) Вторичные(нейрогенные) спинальные и невральные амиотрофии Миастении, миотонии(протекают с нарушением
- 4. МЫШЦЫ ГЛАЗА Круговая мышца глаза
- 5. МЫШЦЫ ГЛАЗА Глазодвигательные мышцы правого глаза
- 7. Обследование пациентов: Генеалогический анализ Неврологический статус Нейроофтальмологическое обследование Электромиография(ЭМГ) Электронейромиография(ЭНМГ) Биохимические исследования Биопсия изменённых мышц с
- 8. ЭМГ и ЭНМГ позволяют судить: О локализации патологического процесса Выявить поражения мышц Поражения нервно-мышечных синапсов Поражения
- 9. Электрофизиологические тесты позволяют легко дифференцировать причину мышечной слабости. Существуют две стандартные методики исследования: ЭМГ, при которой
- 10. ЭЛЕКТРОМИОГРАФИЯ Ребёнок 14 лет, ЭМГ картина в норме
- 11. Для миопатического типа поражения характерно, на ЭМГ: уменьшение средней длительности ПДЕ (на 21 % и более);
- 12. ЭМГ Прогрессирующая мышечная дистрофия(наибольшая выраженность спонтанной активности в виде потенциалов фибрилляций)
- 13. Биохимические исследования Уровень ферментов: Креатинфосфокиназы(КФК общая крови)N=10-110 МЕ Лактатдегидрогеназы(ЛДГ крови)N=0.8-4.0 ммоль/(ч*л) Креатинкреатининовый индекс (Уровень ферментов повышается
- 14. Биопсия изменённых мышц с исследованием биоптата Миопатии и миодистрофии характеризуются диффузной потерей мышечных волокон, которые замещаются
- 15. Прогрессирующие наследственные нейромышечные заболевания Классификация(1974 г. J.Schmitt и J.Renny): 1.Изолированная окулярная миопатия 2.Поздняя окулярная миопатия: офтальмофарингеальная,
- 16. Прогрессирующие наследственные нейромышечные заболевания Окулярная миодистрофия Грефе Офтальмопатия Килона-Невина Окулофарингеальная миодистрофия Бульбарно-паралитическая миопатия Гоффмана Бульбарная амиотрофия
- 17. Наружная прогрессирующая офтальмопатия Килона - Невина Начало заболевания в возрасте до 20 лет, но может встречаться
- 18. Митохондриальная миопатия
- 19. Митохондриальная миопатия
- 20. Окулярная миодистрофия Грефе Наружная хроническая прогрессирующая офтальмоплегия Дебют клинических проявлений в возрасте 20-30 лет. Клиника: *
- 21. Окулофарингеальная миодистрофия Медленное доброкачественное течение, начало заболеваня в возрасте 50 лет. Клиника: * двусторонний парез мышцы,
- 22. Бульбарно-паралитическая миопатия Гоффмана Клиника: *птоз; *парез взора; *лицо миопата; *нарушение глотания, жевания; *дизартрия; *гипотония и гипотрофия
- 23. Бульбарная амиотрофия Фацио-Лонде Вторичная пргрессирующая мышечная дистрофия. Дебютирует в возрасте от 2-12 лет. В оснрве- поражение
- 25. Окулокраниоскелетная прогрессирующая ядерная миопатия Кирнса-Ши Начало заболевания в возрасте до 15 лет. Клиника: *наружный офтальмопарез, постепенно
- 26. МИОИПАТИЯ Офтальмоплегия
- 27. Плече-лопаточно-лицевая миодистрофия Ландузи-Дежерина Чаще встречается у лиц женского пола в возрасте 10-25 лет, медленно прогрессирует. Клиника:
- 28. Офтальмоплегическая прогрессирующая ядерная миопатия с пигментным ретинитом, скротальным языком и снижением интеллекта Клиника: *офтальмоплегия; *значительное снижение
- 29. Невральная амиотрофия Шарко-Мари-Тута-Гоффма Невральная амиотрофия типа А или сочетание признаков наследственной моторной и сенсорной невропатии(НМСН) 1-го
- 30. Офтальмоплегия-плюс, окулокраниосоматическое нейромышечное заболевание, вариант митохондриальной энцефаломиопатии. Болеют чаще лица мужского пола в ювенильном периоде, основные
- 31. Диагностика: Ликвор – белково-клеточная диссоциация; КТ – признаки диффузной гипотрофии головного и спинного мозга; Биохимия крови
- 32. Миастении: Миастения Эрба-Гольдфлама (MIASTENIA GRAVIS) Миастенические кризы Холинергический криз Глазная форма миастении Бульбарная форма миастении Ранняя
- 33. Миастения Эрба-Гольдфлама (MIASTENIA GRAVIS) Хроническое заболевание, проявляющееся слабостью и патологической утомляемостью поперечнополосатых мышц, обусловленное нарушением проведения
- 34. Характерные признаки для мышечной утомляемости при миастении ( М.И. Кузин и Б.М. Гехт, 1996 г.) Избирательное
- 35. Клиника: *птоз верхних век и парезы наружных мышц глаза; *ограничение перемещения взора, страбизм, диплопия; *поражение жевательных,
- 36. Пробы на миастению: Увеличение нагрузки на исследуемые мышцы: например, если 10-20 раз чередовать зажмуривание и максимальное
- 37. Миастения
- 39. Миастенические кризы Выраженные обострения заболевания MIASTENIA GRAVIS. Проявляются нарушением дыхания, сердечно-сосудистыми расстройствами. Экстренная помощь: 1.контроль за
- 41. Холинергический криз Возникает при передозиорвке АХЭП при лечении миастении. Клиника: *миоклонии в мышцах глаз, лица, шеи,
- 42. Глазная форма миастении Клиника: *нарастающий парез мышцы поднимающей верхнее веко; *парез наружных мыщц глаза; *слабость круговой
- 43. Бульбарная форма миастении Клиника: *нарастающая дисфагия; *нарастающее утомление звучности голоса; *слабость мышц шеи, плеч, тазового пояса,
- 44. Ранняя детская форма миастении Проявляется в первые годы жизни. Клиника: *опущение верхних век; *бульбарный синдром; *нарушение
- 45. Юношеская форма миастении Клиника: *проявляется в пубертате, чаще у девочек; *Поражение наружных мышц глаза (птоз, страбизм,
- 46. Врождённая форма миастении Клиника: *чаще встречается у мальчиков; *проявляется при рождении (слабый крик, затруднённое сосание); *птоз
- 47. Миотонии Миолтония Томсена Дистрофичеслая миотония Штейнерта- Баттена- Куршмана Хондродистрофическая миопатия или синдром Шварца- Джампела
- 48. Миотония Томсона Миотонический синдром с рождения или в пубертате. Клиника: *появление миотонии при активных движениях и
- 49. Дистрофичеслая миотония Штейнерта- Баттена- Куршмана Клиника: *начало с ослабления мышечной силы в предплечьях и кистях; *поражение
- 50. Хондродистрофическая миопатия или синдром Шварца- Джампела Клиника: *у детей 3-5 лет; *задержка роста; *дисплазия костей таза;
- 51. Лечение нейромышечных заболеваний: 1. Сбалансированное лечебное питание (продукты, содержащие белок, полиненасыщенные жирные кислоты, витамины, микроэлементы): овощи,
- 53. Скачать презентацию

Нервно-мышечные заболевания – обобщённое название различных видов первичной и вторичной мышечной
Нервно-мышечные заболевания – обобщённое название различных видов первичной и вторичной мышечной

К прогрессирующим мышечным дистрофиям относятся:
Первичные миопатии(амиотрофии)
Вторичные(нейрогенные) спинальные и невральные амиотрофии
Миастении,
К прогрессирующим мышечным дистрофиям относятся:
Первичные миопатии(амиотрофии)
Вторичные(нейрогенные) спинальные и невральные амиотрофии
Миастении,
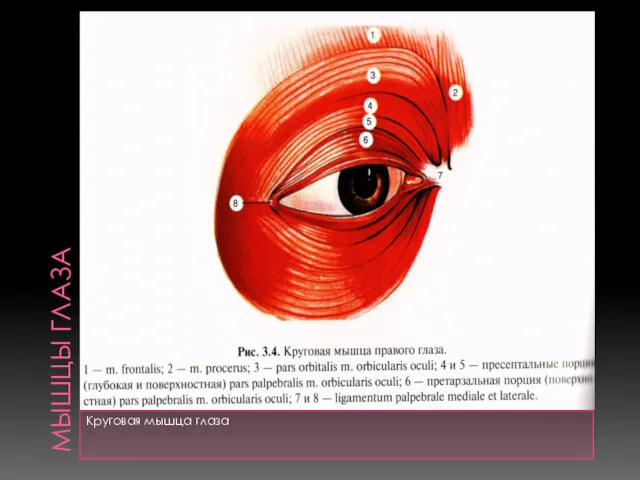
МЫШЦЫ ГЛАЗА
Круговая мышца глаза
МЫШЦЫ ГЛАЗА
Круговая мышца глаза
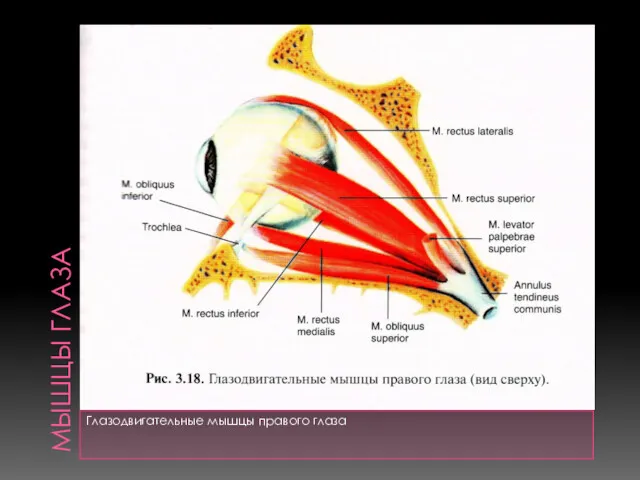
МЫШЦЫ ГЛАЗА
Глазодвигательные мышцы правого глаза
МЫШЦЫ ГЛАЗА
Глазодвигательные мышцы правого глаза


Обследование пациентов:
Генеалогический анализ
Неврологический статус
Нейроофтальмологическое обследование
Электромиография(ЭМГ)
Электронейромиография(ЭНМГ)
Биохимические исследования
Биопсия изменённых мышц с гистологическим изучением
Обследование пациентов:
Генеалогический анализ
Неврологический статус
Нейроофтальмологическое обследование
Электромиография(ЭМГ)
Электронейромиография(ЭНМГ)
Биохимические исследования
Биопсия изменённых мышц с гистологическим изучением

ЭМГ и ЭНМГ позволяют судить:
О локализации патологического процесса
Выявить поражения мышц
Поражения нервно-мышечных
ЭМГ и ЭНМГ позволяют судить:
О локализации патологического процесса
Выявить поражения мышц
Поражения нервно-мышечных

Электрофизиологические тесты позволяют легко дифференцировать причину мышечной слабости.
Существуют две стандартные
Электрофизиологические тесты позволяют легко дифференцировать причину мышечной слабости.
Существуют две стандартные
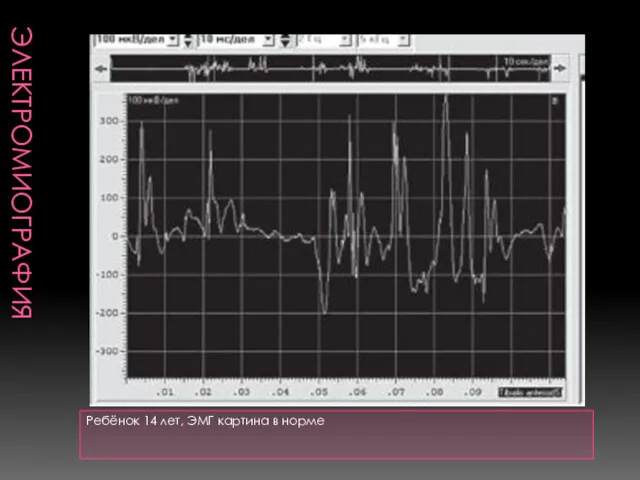
ЭЛЕКТРОМИОГРАФИЯ
Ребёнок 14 лет, ЭМГ картина в норме
ЭЛЕКТРОМИОГРАФИЯ
Ребёнок 14 лет, ЭМГ картина в норме

Для миопатического типа поражения характерно, на ЭМГ:
уменьшение средней длительности ПДЕ (на
Для миопатического типа поражения характерно, на ЭМГ:
уменьшение средней длительности ПДЕ (на
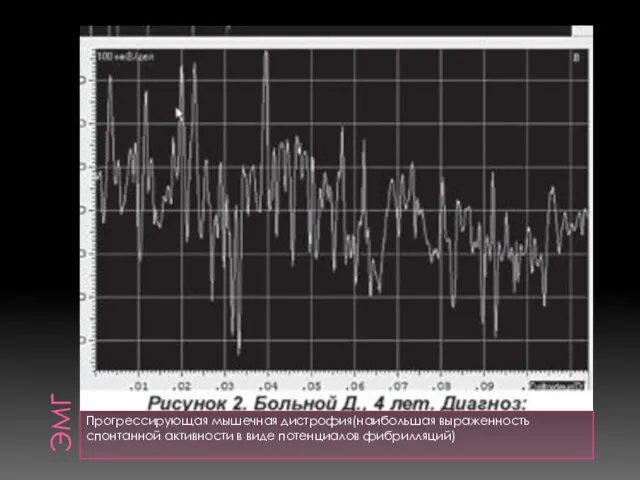
ЭМГ
Прогрессирующая мышечная дистрофия(наибольшая выраженность спонтанной активности в виде потенциалов фибрилляций)
ЭМГ
Прогрессирующая мышечная дистрофия(наибольшая выраженность спонтанной активности в виде потенциалов фибрилляций)

Биохимические исследования
Уровень ферментов:
Креатинфосфокиназы(КФК общая крови)N=10-110 МЕ
Лактатдегидрогеназы(ЛДГ крови)N=0.8-4.0 ммоль/(ч*л)
Креатинкреатининовый индекс (Уровень
Биохимические исследования
Уровень ферментов:
Креатинфосфокиназы(КФК общая крови)N=10-110 МЕ
Лактатдегидрогеназы(ЛДГ крови)N=0.8-4.0 ммоль/(ч*л)
Креатинкреатининовый индекс (Уровень

Биопсия изменённых мышц с исследованием биоптата
Миопатии и миодистрофии характеризуются диффузной
Биопсия изменённых мышц с исследованием биоптата
Миопатии и миодистрофии характеризуются диффузной

Прогрессирующие наследственные нейромышечные заболевания
Классификация(1974 г. J.Schmitt и J.Renny):
1.Изолированная окулярная миопатия
2.Поздняя окулярная
Прогрессирующие наследственные нейромышечные заболевания
Классификация(1974 г. J.Schmitt и J.Renny):
1.Изолированная окулярная миопатия
2.Поздняя окулярная

Прогрессирующие наследственные нейромышечные заболевания
Окулярная миодистрофия Грефе
Офтальмопатия Килона-Невина
Окулофарингеальная миодистрофия
Бульбарно-паралитическая миопатия Гоффмана
Бульбарная амиотрофия
Прогрессирующие наследственные нейромышечные заболевания
Окулярная миодистрофия Грефе
Офтальмопатия Килона-Невина
Окулофарингеальная миодистрофия
Бульбарно-паралитическая миопатия Гоффмана
Бульбарная амиотрофия

Наружная прогрессирующая офтальмопатия Килона - Невина
Начало заболевания в возрасте до 20
Наружная прогрессирующая офтальмопатия Килона - Невина
Начало заболевания в возрасте до 20

Митохондриальная миопатия
Митохондриальная миопатия

Митохондриальная миопатия
Митохондриальная миопатия

Окулярная миодистрофия Грефе
Наружная хроническая прогрессирующая офтальмоплегия
Дебют клинических проявлений в возрасте
Окулярная миодистрофия Грефе
Наружная хроническая прогрессирующая офтальмоплегия
Дебют клинических проявлений в возрасте

Окулофарингеальная миодистрофия
Медленное доброкачественное течение, начало заболеваня в возрасте 50 лет.
Клиника:
*
Окулофарингеальная миодистрофия
Медленное доброкачественное течение, начало заболеваня в возрасте 50 лет.
Клиника:
*

Бульбарно-паралитическая миопатия Гоффмана
Клиника:
*птоз;
*парез взора;
*лицо миопата;
*нарушение глотания, жевания;
*дизартрия;
*гипотония и гипотрофия мышц плечевого
Бульбарно-паралитическая миопатия Гоффмана
Клиника:
*птоз;
*парез взора;
*лицо миопата;
*нарушение глотания, жевания;
*дизартрия;
*гипотония и гипотрофия мышц плечевого

Бульбарная амиотрофия Фацио-Лонде
Вторичная пргрессирующая мышечная дистрофия.
Дебютирует в возрасте от 2-12 лет.
В
Бульбарная амиотрофия Фацио-Лонде
Вторичная пргрессирующая мышечная дистрофия.
Дебютирует в возрасте от 2-12 лет.
В


Окулокраниоскелетная прогрессирующая ядерная миопатия Кирнса-Ши
Начало заболевания в возрасте до 15 лет.
Клиника:
*наружный
Окулокраниоскелетная прогрессирующая ядерная миопатия Кирнса-Ши
Начало заболевания в возрасте до 15 лет.
Клиника:
*наружный
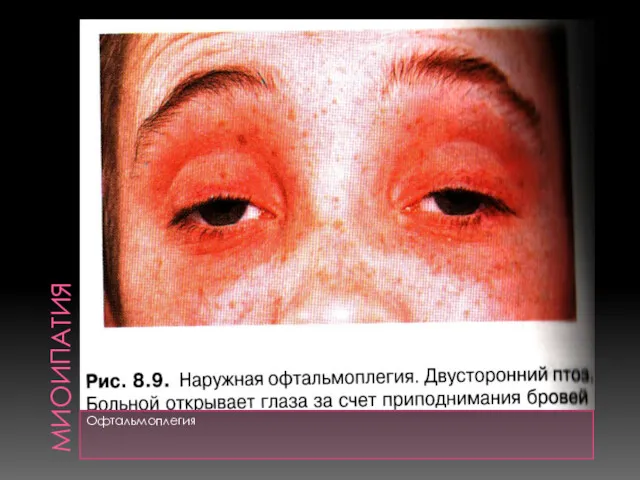
МИОИПАТИЯ
Офтальмоплегия
МИОИПАТИЯ
Офтальмоплегия

Плече-лопаточно-лицевая миодистрофия Ландузи-Дежерина
Чаще встречается у лиц женского пола в возрасте 10-25
Плече-лопаточно-лицевая миодистрофия Ландузи-Дежерина
Чаще встречается у лиц женского пола в возрасте 10-25

Офтальмоплегическая прогрессирующая ядерная миопатия с пигментным ретинитом, скротальным языком и снижением
Офтальмоплегическая прогрессирующая ядерная миопатия с пигментным ретинитом, скротальным языком и снижением

Невральная амиотрофия Шарко-Мари-Тута-Гоффма
Невральная амиотрофия типа А или сочетание признаков наследственной моторной
Невральная амиотрофия Шарко-Мари-Тута-Гоффма
Невральная амиотрофия типа А или сочетание признаков наследственной моторной

Офтальмоплегия-плюс, окулокраниосоматическое нейромышечное заболевание, вариант митохондриальной энцефаломиопатии.
Болеют чаще лица мужского пола
Офтальмоплегия-плюс, окулокраниосоматическое нейромышечное заболевание, вариант митохондриальной энцефаломиопатии.
Болеют чаще лица мужского пола

Диагностика:
Ликвор – белково-клеточная диссоциация;
КТ – признаки диффузной гипотрофии головного и спинного
Диагностика:
Ликвор – белково-клеточная диссоциация;
КТ – признаки диффузной гипотрофии головного и спинного

Миастении:
Миастения Эрба-Гольдфлама (MIASTENIA GRAVIS)
Миастенические кризы
Холинергический криз
Глазная форма миастении
Бульбарная форма миастении
Ранняя детская
Миастении:
Миастения Эрба-Гольдфлама (MIASTENIA GRAVIS)
Миастенические кризы
Холинергический криз
Глазная форма миастении
Бульбарная форма миастении
Ранняя детская

Миастения Эрба-Гольдфлама (MIASTENIA GRAVIS)
Хроническое заболевание, проявляющееся слабостью и патологической утомляемостью поперечнополосатых
Миастения Эрба-Гольдфлама (MIASTENIA GRAVIS)
Хроническое заболевание, проявляющееся слабостью и патологической утомляемостью поперечнополосатых

Характерные признаки для мышечной утомляемости при миастении ( М.И. Кузин и
Характерные признаки для мышечной утомляемости при миастении ( М.И. Кузин и

Клиника:
*птоз верхних век и парезы наружных мышц глаза;
*ограничение перемещения взора, страбизм,
Клиника:
*птоз верхних век и парезы наружных мышц глаза;
*ограничение перемещения взора, страбизм,

Пробы на миастению:
Увеличение нагрузки на исследуемые мышцы: например, если 10-20 раз
Пробы на миастению:
Увеличение нагрузки на исследуемые мышцы: например, если 10-20 раз
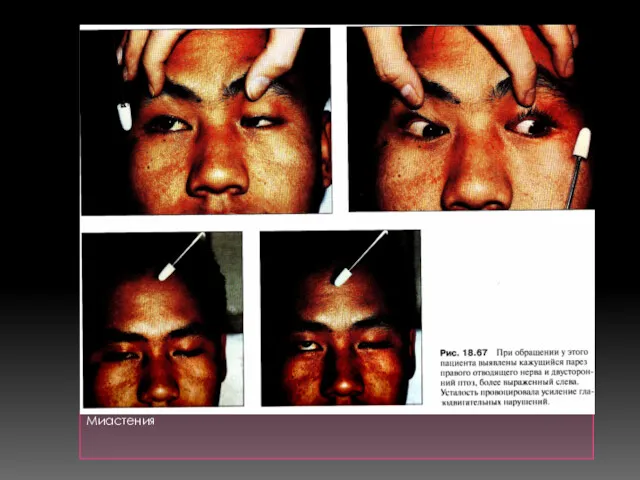
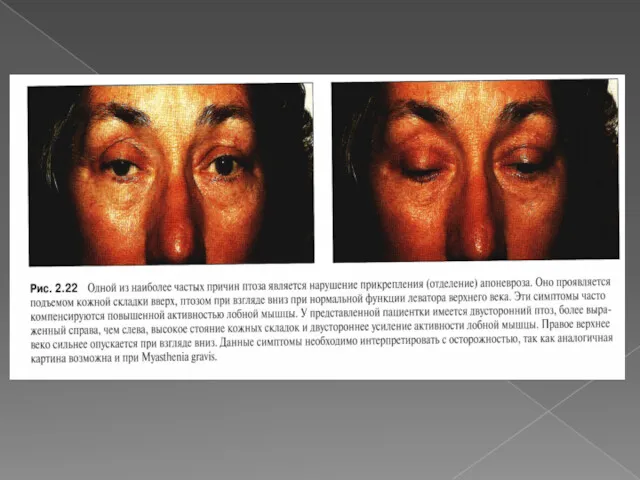
Миастения
Миастения

Миастенические кризы
Выраженные обострения заболевания MIASTENIA GRAVIS.
Проявляются нарушением дыхания, сердечно-сосудистыми расстройствами.
Экстренная помощь:
Миастенические кризы
Выраженные обострения заболевания MIASTENIA GRAVIS.
Проявляются нарушением дыхания, сердечно-сосудистыми расстройствами.
Экстренная помощь:


Холинергический криз
Возникает при передозиорвке АХЭП при лечении миастении.
Клиника:
*миоклонии в мышцах глаз,
Холинергический криз
Возникает при передозиорвке АХЭП при лечении миастении.
Клиника:
*миоклонии в мышцах глаз,

Глазная форма миастении
Клиника:
*нарастающий парез мышцы поднимающей верхнее веко;
*парез наружных мыщц глаза;
*слабость
Глазная форма миастении
Клиника:
*нарастающий парез мышцы поднимающей верхнее веко;
*парез наружных мыщц глаза;
*слабость

Бульбарная форма миастении
Клиника:
*нарастающая дисфагия;
*нарастающее утомление звучности голоса;
*слабость мышц шеи, плеч, тазового
Бульбарная форма миастении
Клиника:
*нарастающая дисфагия;
*нарастающее утомление звучности голоса;
*слабость мышц шеи, плеч, тазового

Ранняя детская форма миастении
Проявляется в первые годы жизни.
Клиника:
*опущение верхних век;
*бульбарный
Ранняя детская форма миастении
Проявляется в первые годы жизни.
Клиника:
*опущение верхних век;
*бульбарный

Юношеская форма миастении
Клиника:
*проявляется в пубертате, чаще у девочек;
*Поражение наружных мышц глаза
Юношеская форма миастении
Клиника:
*проявляется в пубертате, чаще у девочек;
*Поражение наружных мышц глаза

Врождённая форма миастении
Клиника:
*чаще встречается у мальчиков;
*проявляется при рождении (слабый крик,
Врождённая форма миастении
Клиника:
*чаще встречается у мальчиков;
*проявляется при рождении (слабый крик,

Миотонии
Миолтония Томсена
Дистрофичеслая миотония Штейнерта- Баттена- Куршмана
Хондродистрофическая миопатия или синдром Шварца- Джампела
Миотонии
Миолтония Томсена
Дистрофичеслая миотония Штейнерта- Баттена- Куршмана
Хондродистрофическая миопатия или синдром Шварца- Джампела

Миотония Томсона
Миотонический синдром с рождения или в пубертате.
Клиника:
*появление миотонии при активных
Миотония Томсона
Миотонический синдром с рождения или в пубертате.
Клиника:
*появление миотонии при активных

Дистрофичеслая миотония Штейнерта- Баттена- Куршмана
Клиника:
*начало с ослабления мышечной силы в предплечьях
Дистрофичеслая миотония Штейнерта- Баттена- Куршмана
Клиника:
*начало с ослабления мышечной силы в предплечьях

Хондродистрофическая миопатия или синдром Шварца- Джампела
Клиника:
*у детей 3-5 лет;
*задержка роста;
*дисплазия костей
Хондродистрофическая миопатия или синдром Шварца- Джампела
Клиника:
*у детей 3-5 лет;
*задержка роста;
*дисплазия костей

Лечение нейромышечных заболеваний:
1. Сбалансированное лечебное питание (продукты, содержащие белок, полиненасыщенные
Лечение нейромышечных заболеваний:
1. Сбалансированное лечебное питание (продукты, содержащие белок, полиненасыщенные
 Ранний послеоперационный период
Ранний послеоперационный период Соціальна медицина та організація охорони здоров’я як наука. Предмет, методи, значення для практики охорони здоров’я
Соціальна медицина та організація охорони здоров’я як наука. Предмет, методи, значення для практики охорони здоров’я Физиологические механизмы регуляции в организме
Физиологические механизмы регуляции в организме Герпетическая инфекция
Герпетическая инфекция Аналық бездің қатерлі ісігі
Аналық бездің қатерлі ісігі Обследование альвеолярного отростка верхней и нижней челюсти
Обследование альвеолярного отростка верхней и нижней челюсти Одонтогенные новообразования челюстей у детей (амелобластома, одонтома, цементома)
Одонтогенные новообразования челюстей у детей (амелобластома, одонтома, цементома) Малярия, токсоплазма, лямблии
Малярия, токсоплазма, лямблии Балалар жақ сүйегінің периоститі
Балалар жақ сүйегінің периоститі Патологическая стираемость твердых тканей зубов
Патологическая стираемость твердых тканей зубов Балаларда туа пайда болған жүрек ақаулары
Балаларда туа пайда болған жүрек ақаулары Скажи наркотикам нет
Скажи наркотикам нет Оптимизация обучения студентов медицинского университета
Оптимизация обучения студентов медицинского университета Жасқа байланысты,асқынбаған тіс жегісінде,жүйелі аурулар мен зат алмасу ауруларында ұлпадағы өзгерістер
Жасқа байланысты,асқынбаған тіс жегісінде,жүйелі аурулар мен зат алмасу ауруларында ұлпадағы өзгерістер Предварительные и периодические медицинские осмотры работников
Предварительные и периодические медицинские осмотры работников Применения Ботулинотоксин-А при лечении невралгии тройничного нерва
Применения Ботулинотоксин-А при лечении невралгии тройничного нерва Әйел жыныс мүшелерінің қабыну аурулары
Әйел жыныс мүшелерінің қабыну аурулары Метаболизм липидов
Метаболизм липидов Рак поджелудочной железы
Рак поджелудочной железы Учение о диагнозе
Учение о диагнозе Введение в сексологию
Введение в сексологию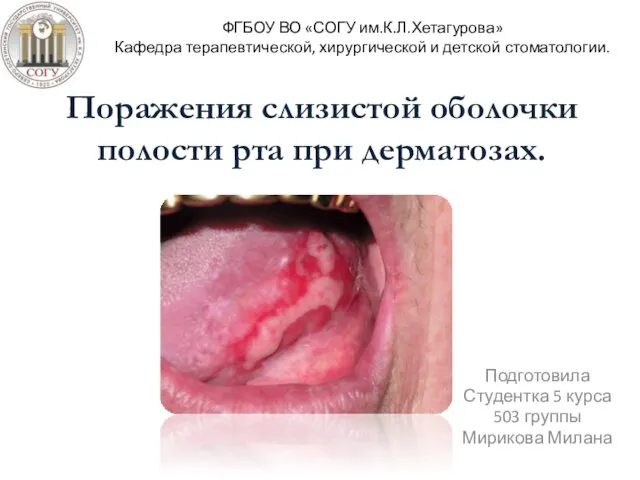 Поражения слизистой оболочки полости рта при дерматозах
Поражения слизистой оболочки полости рта при дерматозах Биохимия слюны
Биохимия слюны Профилактика наркомании и формирование установок на ведение здорового образа жизни среди молодежи
Профилактика наркомании и формирование установок на ведение здорового образа жизни среди молодежи Актуальные вопросы проведения химиопрофилактики перинатальной передачи ВИЧ от матери ребенку в Алтайском крае
Актуальные вопросы проведения химиопрофилактики перинатальной передачи ВИЧ от матери ребенку в Алтайском крае Диагностика раннего рака желудка
Диагностика раннего рака желудка Modal Verbs Lobular Pneumonia
Modal Verbs Lobular Pneumonia Аутоиммунный гепатит. Определение. Классификация
Аутоиммунный гепатит. Определение. Классификация