- Патофизиология сахарного диабета

Содержание
- 2. Diabetes (греч.) - проходить сквозь Сахарный диабет – это заболевание, основным патогенетическим фактором в патогенезе которого
- 3. Этиология и патогенез Ведущим патогенетическим фактором в развитии сахарного диабета является инсулиновая недостаточность.
- 4. Существуют 2 формы инсулиновой недостаточности: панкреатическая и внепанкреатическая. Панкреатическая форма инсулиновой недостаточности характеризуется абсолютной инсулиновой недостаточностью,
- 5. Для ΙΙ типа СД (ИНСД) характерна инсулинорезистентность - это снижение реакции инсулиночувствительных тканей на инсулин при
- 6. Причины сахарного диабета 1 типа 1.Кислородное голодание ткани железы (атеросклероз, спазм сосудов, кровоизлияние и т.д.) –
- 7. 3. Истощение β- клеток островков (перенапряжение) 3.1. Алиментарный фактор - при излишнем употреблении в пищу легкоусвояемых
- 8. 5.Нарушение пуринового обмена - при образовании в организме аллоксана, близкого по структуре к мочевой кислоте (уреид
- 9. Причины сахарного диабета 2 типа 1.Избыточная продукция контринсулярных гормонов: СТГ, глюкокортикоидов, адреналина 2.Повышенная активность фермента инсулиназы
- 10. 4.Нарушение в гормональном рецепторе 4.1. Генетические: - нарушение синтеза субстрата инсулинового рецептора; - нарушение встраивания рецептора
- 11. 5.Нарушение связывания гормонов белками крови – увеличение инсулина в связанной с белком форме. Инсулин в прочной
- 12. Отличия ИЗСД (I типа) и ИНСД (II тип)
- 13. Отличия ИЗСД (I типа) и ИНСД (II тип)
- 14. В мире каждый час совершается 55 ампутаций нижних конечностей у больных СД, в нашей стране –
- 15. Нарушения обмена веществ 1. Углеводного 2. Липидного 3. Белкового 4. Водно-солевого
- 16. ГЛЮКОЗА 3,33 - 5,55 ммоль/л HB A1c (%) 4 - 6% ( Нарушения углеводного обмена 1.
- 17. Ранним признаком нарушения углеводного обмена является колебание глюкозы в крови натощак. При этом происходит изменение осмотических
- 18. Глюкозотолерантный тест – оценка углеводного обмена, основанная на определении уровня глюкозы в крови натощак и после
- 19. Показатели глюкозотолерантного теста (ммоль/л)
- 20. САХАРНЫЕ КРИВЫЕ 1- здорового человека 2- при нарушен- ной толерантно- сти к глюкозе 3- при сахарном
- 21. В норме уровень глюкозы в крови при этом тесте достигает максимума через 30 минут, его величина
- 22. Нарушенная толерантность к глюкозе с кривыми, располагающимися между нормальной и диабетической, может наблюдаться у людей, которые
- 23. Важным критерием для выявления инсулиновой недостаточности служит определение содержания глюкозы в плазме крови натощак. В норме
- 24. ЗНАЧЕНИЕ ГИПЕРГЛИКЕМИИ В ПАТОГЕНЕЗЕ САХАРНОГО ДИАБЕТА Гипергликемия играет саногенетическую роль: Гипергликемия является признаком нарушения углеводного обмена.
- 25. Патогенетическая роль гипергликемии при сахарном диабете заключается в том, что увеличивается поступление глюкозы в клетки инсулинонезависимых
- 26. При этом усиливаются процессы превращения глюкозы в осмотически активные вещества (сорбитол, фруктоза). В клетках увеличивается концентрация
- 27. Также гипергликемия оказывает токсический эффект продуктами гликирования (молочная, пировиноградная кислоты), сопровождается гиперосмолярностью плазмы крови, что приводит
- 28. Гликированный гемоглобин - образуется при неферментативном гликозилировании гемоглобина. Определение гликированного гемоглобина имеет большое диагностическое значение, это
- 29. Основным методом профилактического контроля гликемии является поддержание нормального уровня гликированного гемоглобина (меньше 6,5%) на протяжении 5-10
- 30. Причинами глюкозурии при сахарном диабете являются: 1) гипергликемия, превышающая почечный порог (9,9 ммоль/л), то есть максимальную
- 31. Терапевтические цели по гликемии при СД. (Федеральная Целевая Программа «Сахарный диабет»)
- 32. Нарушения углеводного обмена лежат в основе кардиоваскулярной патологии с возникновением дисфункции эндотелия.
- 33. Нарушения жирового обмена 1. Гиперлипидемия ( > СЖК) 2. Жировая инфильтрация печени 3. Гиперкетонемия 4. Гиперхолестеринемия
- 34. Гиперлипидемия необходима для обеспечения организма энергией в условиях снижения утилизации глюкозы. Гиперлипидемия связана с возрастанием липолиза
- 35. Следующим признаком нарушения жирового обмена является гиперкетонемия. Печень переключает метаболизм поступающих в избытке свободных жирных кислот
- 36. При окислении жирных кислот образуется большое количество ацетил-коэнзимА, который в условиях торможения липогенеза (из-за дефицита НАДФ•Н
- 37. В условиях избытка образования ацетил-коэнзимА и ацетоуксусной кислоты усиливается синтез холестерина, ЛПОНП и ЛПНП. Гиперхолестеринемия является
- 38. Следующим признаком нарушения липидного обмена при сахарном диабете является кетонурия. Кетоновые тела выводятся с мочой, что
- 39. Нарушения белкового обмена уменьшение синтеза белка, увеличение распада белка, гипо- и парапротеинемия, снижение антителообразования, ослабление иммунитета,
- 40. Уменьшение синтеза белка обусловлено - уменьшением проницаемости клеточных мембран для аминокислот (инсулинозависимый процесс); - недостаточным обеспечением
- 41. активацией глюконеогенеза (в особенности в мышечной ткани), при этом в крови и моче регистрируется возрастание уровней
- 42. Избыточный катаболизм белка и сниженный синтез затрудняют нормальное течение регенераторных процессов, с чем связывается плохое заживление
- 43. Нарушение водно-электролитного баланса При СД возникает полиурия (выделение мочи более 2 л/сутки) и полидипсия. Полиурия возникает
- 44. Патогенез острых и хронических осложнений сахарного диабета При СД выделяют две группы осложнений: острые и хронические.
- 45. Осложнения сахарного диабета Острые Хронические - Гиперкетонемическая кома - Гиперосмолярная кома - Гиперлактацидемическая кома - Гипогликемическая
- 46. Осложнения сахарного диабета СД I типа СД II типа - микроангиопатии - макроангиопатии - ретинопатии -
- 47. КОМЫ 1. Кетоацидотическая 2. Гиперосмолярная 3. Гиперлактацидемическая 4. Гипогликемическая
- 48. Кетоацидотическая (истинная) кома Патогенез: Клиника: - гиперкетонемия - начало постепенное - метаболический ацидоз (полидипсия, полиурия, слабость,
- 49. Развитие комы прежде всего связано с токсическим влиянием на ЦНС продуктов нарушенного обмена. Кетоацидотическая кома, как
- 50. В механизме кетоацидотической комы немаловажное значение имеет метаболический ацидоз, связанный с накоплением кислых продуктов нарушенных обменов.
- 51. Гиперосмолярная кома Причины: - типична для СД II - может быть без СД в анамнезе -
- 52. Данная кома характеризуется резко выраженными признаками: гиперосмолярность, гипергликемия и дегидратация. Чаще всего бывает без кетоацидоза, так
- 53. Дегидратация структур головного мозга с резким падением внутричерепного давления приводит к общему угнетению центральной нервной системы,
- 54. Гиперлактацидемическая кома Причины: - встречается при СД I и II - в условиях гипоксии Патогенез: -
- 55. Гиперлактацидемическая кома развивается быстро, обычно в течение нескольких часов. При осмотре больного признаки дегидратации выражены меньше
- 56. Гипогликемическая кома Причины: - передозировка сахаросни- жающими препаратами у больных СД II - гиперинсулинизм Патогенез: -
- 57. Причиной гипогликемической комы является абсолютная недостаточность глюкозы для обеспечения энергетических процессов в нейронах центральной нервной системы.
- 58. Хронические осложнения сахарного диабета К хроническим осложнениям СД относятся: ангиопатии (микроангиопатии; макроангиопатии) полинейропатии кардиоваскулярная патология артериальная
- 59. 1. Неферментативное гликозилирование белков базальных мембран капилляров, приводящее: а) к «сшивке» коллагена базальной мембраны сосудов с
- 60. Последствия микроангиопатии Набухание, утолщение и дистрофия эндотелия сосудов. Изменение строения белков базальной мембраны сосудов и приобретение
- 61. 1 Увеличение концентрации глико- и мукопротеидов и отложением гиалина в базальных мембранах и интерстиции сосудов. 2
- 62. 8 Накопление сорбитола в стенке артериальных сосудов. 9 Активация синтеза тромбоксана А2 тромбоцитами, что потенцирует вазоконстрикцию
- 63. КАРДИОВАСКУЛЯРНАЯ ПАТОЛОГИЯ является основной причиной, вызывающей высокую летальность у больных сахарным диабетом. Выделяют: - нарушение деятельности
- 64. Патогенез нарушений деятельности сердца - возникновение дистрофических изменений в миокарде за счет а) нарушения электролитного баланса,
- 65. Причины: Гиперинсулинемия способствует задержке в организме Na. При этом Na потенцирует действие катехоламинов. 2. Инсулин снижает
- 66. Основные звенья патогенеза диабетической нейропатии: Снижение интраневрального кровоснабжения в связи с развитием хронической ишемии и гипоксии
- 67. Патогенез нефропатий Нарушение функций почек - одна из частых причин инвалидизации и смерти при СД. Причины:
- 68. Ретинопатия Поражение сетчатки глаза при диабете выявляют примерно у 3% больных в дебюте заболевания, более чем
- 69. Ретинопатия
- 70. Диабетическая стопа Факторы риска: длительность сахарного диабета более 10 лет; возраст более 40 лет; атеросклероз артерий
- 71. Формы: Нейропатическая (язвы, остеоартропатии, отеки) Ишемическая (гангрена) Диабетическая стопа
- 72. Патогенез: - ишемия (следствие микро-, макроангиопатий), - нарушение трофики (следствие диабетической полинейропатии), - инфекция, - деструкция
- 74. Показатели контроля компенсации СД
- 75. 1. Этиологический – направлен на устранение причины на начальном этапе 2. Патогенетический – направлен на разрыв
- 76. В терапии СД 2 типа в последние годы уделяют большое внимание гормону инкретину, который продуцируется в
- 77. Воспроизведение сахарного диабета в эксперименте Основные сведения об этиологии и патогенезе сахарного диабета стали известны благодаря
- 78. Широкое распространение получила модель аллоксанового диабета, возникающего при введении животным аллоксана. Это вещество избирательно повреждает β-клетки
- 80. Скачать презентацию

Diabetes (греч.) - проходить сквозь
Сахарный диабет – это заболевание, основным патогенетическим
Сахарный диабет – это заболевание, основным патогенетическим

Этиология и патогенез
Ведущим патогенетическим фактором в развитии сахарного диабета является инсулиновая
Ведущим патогенетическим фактором в развитии сахарного диабета является инсулиновая

Существуют 2 формы инсулиновой недостаточности: панкреатическая и внепанкреатическая.
Панкреатическая форма инсулиновой
Существуют 2 формы инсулиновой недостаточности: панкреатическая и внепанкреатическая.
Панкреатическая форма инсулиновой

Для ΙΙ типа СД (ИНСД) характерна инсулинорезистентность - это снижение реакции

Причины сахарного диабета 1 типа
1.Кислородное голодание ткани железы (атеросклероз, спазм сосудов,
Причины сахарного диабета 1 типа
1.Кислородное голодание ткани железы (атеросклероз, спазм сосудов,

3. Истощение β- клеток островков (перенапряжение)
3.1. Алиментарный фактор - при излишнем
3. Истощение β- клеток островков (перенапряжение)
3.1. Алиментарный фактор - при излишнем

5.Нарушение пуринового обмена - при образовании в организме аллоксана, близкого по
5.Нарушение пуринового обмена - при образовании в организме аллоксана, близкого по

Причины сахарного диабета 2 типа
1.Избыточная продукция контринсулярных гормонов: СТГ, глюкокортикоидов, адреналина
2.Повышенная
Причины сахарного диабета 2 типа
1.Избыточная продукция контринсулярных гормонов: СТГ, глюкокортикоидов, адреналина
2.Повышенная

4.Нарушение в гормональном рецепторе
4.1. Генетические:
- нарушение синтеза субстрата инсулинового рецептора;
4.Нарушение в гормональном рецепторе
4.1. Генетические:
- нарушение синтеза субстрата инсулинового рецептора;

5.Нарушение связывания гормонов белками крови – увеличение инсулина в связанной с
5.Нарушение связывания гормонов белками крови – увеличение инсулина в связанной с

Отличия ИЗСД (I типа) и ИНСД (II тип)
Отличия ИЗСД (I типа) и ИНСД (II тип)

Отличия ИЗСД (I типа) и ИНСД (II тип)
Отличия ИЗСД (I типа) и ИНСД (II тип)

В мире каждый час совершается 55 ампутаций нижних конечностей у
В мире каждый час совершается 55 ампутаций нижних конечностей у

Нарушения обмена веществ
1. Углеводного
2. Липидного
3. Белкового
4. Водно-солевого
1. Углеводного
2. Липидного
3. Белкового
4. Водно-солевого

ГЛЮКОЗА 3,33 - 5,55 ммоль/л
HB A1c (%) 4 -
ГЛЮКОЗА 3,33 - 5,55 ммоль/л
HB A1c (%) 4 -

Ранним признаком нарушения углеводного обмена является колебание глюкозы в крови натощак.
Ранним признаком нарушения углеводного обмена является колебание глюкозы в крови натощак.

Глюкозотолерантный тест
– оценка углеводного обмена, основанная на определении уровня глюкозы
Глюкозотолерантный тест
– оценка углеводного обмена, основанная на определении уровня глюкозы

Показатели глюкозотолерантного теста
(ммоль/л)
Показатели глюкозотолерантного теста
(ммоль/л)
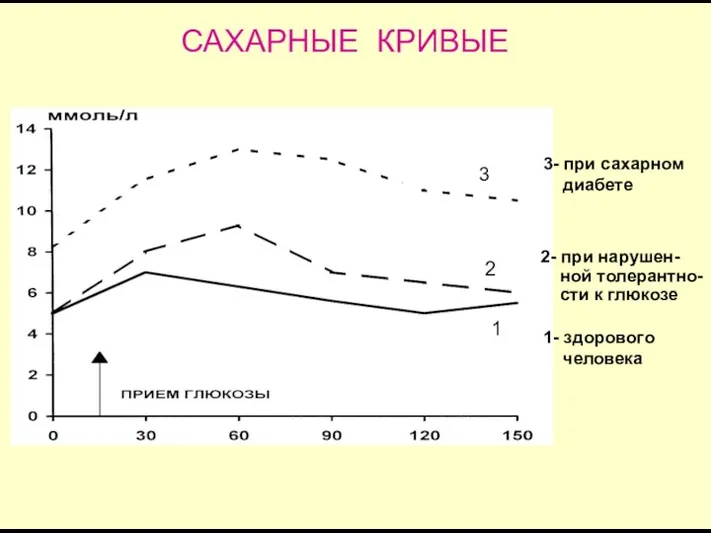
САХАРНЫЕ КРИВЫЕ
1- здорового
человека
2- при нарушен-
ной толерантно-
сти к глюкозе
3-
САХАРНЫЕ КРИВЫЕ
1- здорового
человека
2- при нарушен-
ной толерантно-
сти к глюкозе
3-

В норме уровень глюкозы в крови при этом тесте достигает максимума
В норме уровень глюкозы в крови при этом тесте достигает максимума

Нарушенная толерантность к глюкозе с кривыми, располагающимися между нормальной и диабетической,
Нарушенная толерантность к глюкозе с кривыми, располагающимися между нормальной и диабетической,

Важным критерием для выявления инсулиновой недостаточности служит определение содержания глюкозы в
Важным критерием для выявления инсулиновой недостаточности служит определение содержания глюкозы в

ЗНАЧЕНИЕ ГИПЕРГЛИКЕМИИ В ПАТОГЕНЕЗЕ САХАРНОГО ДИАБЕТА
Гипергликемия играет саногенетическую роль:
Гипергликемия является признаком
ЗНАЧЕНИЕ ГИПЕРГЛИКЕМИИ В ПАТОГЕНЕЗЕ САХАРНОГО ДИАБЕТА
Гипергликемия играет саногенетическую роль:
Гипергликемия является признаком

Патогенетическая роль гипергликемии при сахарном диабете заключается в том, что увеличивается
Патогенетическая роль гипергликемии при сахарном диабете заключается в том, что увеличивается

При этом усиливаются процессы превращения глюкозы в осмотически активные вещества (сорбитол,
При этом усиливаются процессы превращения глюкозы в осмотически активные вещества (сорбитол,

Также гипергликемия оказывает токсический эффект продуктами гликирования (молочная, пировиноградная кислоты),
сопровождается гиперосмолярностью
Также гипергликемия оказывает токсический эффект продуктами гликирования (молочная, пировиноградная кислоты),
сопровождается гиперосмолярностью

Гликированный гемоглобин
- образуется при неферментативном гликозилировании гемоглобина. Определение гликированного
Гликированный гемоглобин
- образуется при неферментативном гликозилировании гемоглобина. Определение гликированного

Основным методом профилактического контроля гликемии является поддержание нормального уровня гликированного гемоглобина
Основным методом профилактического контроля гликемии является поддержание нормального уровня гликированного гемоглобина

Причинами глюкозурии
при сахарном диабете являются:
1) гипергликемия, превышающая почечный порог
Причинами глюкозурии
при сахарном диабете являются:
1) гипергликемия, превышающая почечный порог
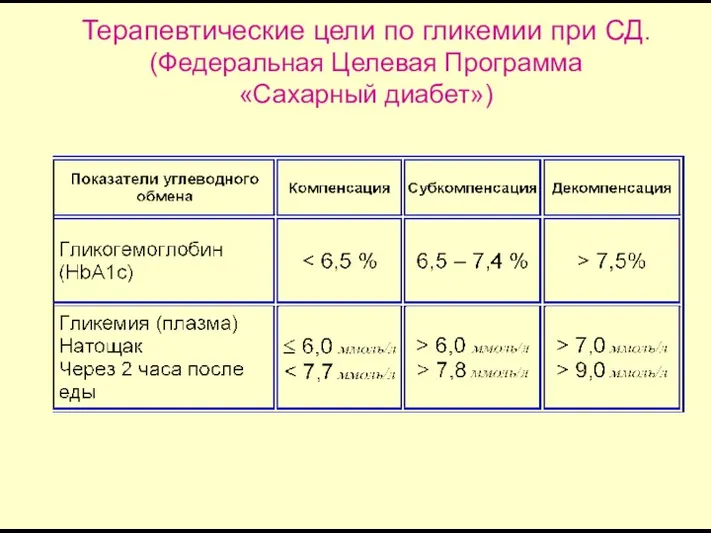
Терапевтические цели по гликемии при СД.
(Федеральная Целевая Программа
«Сахарный диабет»)
Терапевтические цели по гликемии при СД.
(Федеральная Целевая Программа
«Сахарный диабет»)

Нарушения углеводного обмена лежат в основе кардиоваскулярной патологии с возникновением дисфункции
Нарушения углеводного обмена лежат в основе кардиоваскулярной патологии с возникновением дисфункции

Нарушения жирового обмена
1. Гиперлипидемия ( > СЖК)
2. Жировая инфильтрация печени
3. Гиперкетонемия
4.
1. Гиперлипидемия ( > СЖК)
2. Жировая инфильтрация печени
3. Гиперкетонемия
4.

Гиперлипидемия необходима для обеспечения организма энергией в условиях снижения утилизации глюкозы.
Гиперлипидемия необходима для обеспечения организма энергией в условиях снижения утилизации глюкозы.

Следующим признаком нарушения жирового обмена является гиперкетонемия. Печень переключает метаболизм поступающих
Следующим признаком нарушения жирового обмена является гиперкетонемия. Печень переключает метаболизм поступающих

При окислении жирных кислот образуется большое количество ацетил-коэнзимА, который в условиях
При окислении жирных кислот образуется большое количество ацетил-коэнзимА, который в условиях

В условиях избытка образования ацетил-коэнзимА и ацетоуксусной кислоты усиливается синтез холестерина,
В условиях избытка образования ацетил-коэнзимА и ацетоуксусной кислоты усиливается синтез холестерина,

Следующим признаком нарушения липидного обмена при сахарном диабете является кетонурия. Кетоновые
Следующим признаком нарушения липидного обмена при сахарном диабете является кетонурия. Кетоновые

Нарушения белкового обмена
уменьшение синтеза белка,
увеличение распада белка,
гипо- и парапротеинемия,
снижение
Нарушения белкового обмена
уменьшение синтеза белка,
увеличение распада белка,
гипо- и парапротеинемия,
снижение

Уменьшение синтеза белка обусловлено
- уменьшением проницаемости клеточных мембран для аминокислот (инсулинозависимый
Уменьшение синтеза белка обусловлено
- уменьшением проницаемости клеточных мембран для аминокислот (инсулинозависимый

активацией глюконеогенеза (в особенности в мышечной ткани), при этом в крови
активацией глюконеогенеза (в особенности в мышечной ткани), при этом в крови

Избыточный катаболизм белка и сниженный синтез затрудняют нормальное течение регенераторных процессов,
Избыточный катаболизм белка и сниженный синтез затрудняют нормальное течение регенераторных процессов,

Нарушение водно-электролитного баланса
При СД возникает полиурия (выделение мочи более 2 л/сутки)
Нарушение водно-электролитного баланса
При СД возникает полиурия (выделение мочи более 2 л/сутки)

Патогенез острых и хронических
осложнений сахарного диабета
При СД выделяют две группы
Патогенез острых и хронических
осложнений сахарного диабета
При СД выделяют две группы
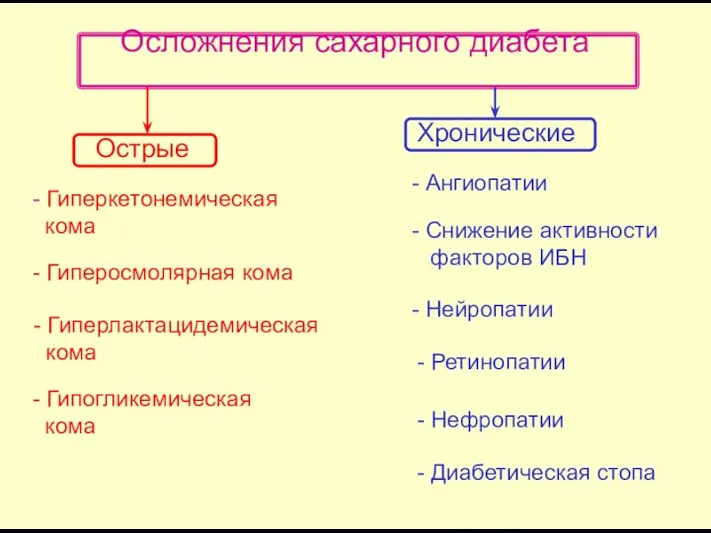
Осложнения сахарного диабета
Острые
Хронические
- Гиперкетонемическая
кома
- Гиперосмолярная кома
- Гиперлактацидемическая
кома
- Гипогликемическая
кома
-
Осложнения сахарного диабета
Острые
Хронические
- Гиперкетонемическая
кома
- Гиперосмолярная кома
- Гиперлактацидемическая
кома
- Гипогликемическая
кома
-

Осложнения сахарного диабета
СД I типа СД II типа
- микроангиопатии -
Осложнения сахарного диабета
СД I типа СД II типа
- микроангиопатии -

КОМЫ
1. Кетоацидотическая
2. Гиперосмолярная
3. Гиперлактацидемическая
4. Гипогликемическая
КОМЫ
1. Кетоацидотическая
2. Гиперосмолярная
3. Гиперлактацидемическая
4. Гипогликемическая

Кетоацидотическая (истинная) кома
Патогенез: Клиника:
- гиперкетонемия - начало постепенное
- метаболический
Кетоацидотическая (истинная) кома
Патогенез: Клиника:
- гиперкетонемия - начало постепенное
- метаболический

Развитие комы прежде всего связано с токсическим влиянием на ЦНС продуктов
Развитие комы прежде всего связано с токсическим влиянием на ЦНС продуктов

В механизме кетоацидотической комы немаловажное значение имеет метаболический ацидоз, связанный с
В механизме кетоацидотической комы немаловажное значение имеет метаболический ацидоз, связанный с

Гиперосмолярная кома
Причины:
- типична для СД II
- может быть без СД в
Гиперосмолярная кома
Причины:
- типична для СД II
- может быть без СД в

Данная кома характеризуется резко выраженными признаками: гиперосмолярность, гипергликемия и дегидратация.
Чаще
Данная кома характеризуется резко выраженными признаками: гиперосмолярность, гипергликемия и дегидратация.
Чаще

Дегидратация структур головного мозга с резким падением внутричерепного давления приводит к
Дегидратация структур головного мозга с резким падением внутричерепного давления приводит к

Гиперлактацидемическая кома
Причины:
- встречается при СД I и II
- в условиях гипоксии
Патогенез:
-
Гиперлактацидемическая кома
Причины:
- встречается при СД I и II
- в условиях гипоксии
Патогенез:
-

Гиперлактацидемическая кома развивается быстро, обычно в течение нескольких часов. При осмотре
Гиперлактацидемическая кома развивается быстро, обычно в течение нескольких часов. При осмотре

Гипогликемическая кома
Причины:
- передозировка сахаросни-
жающими препаратами у
больных СД II
-
Гипогликемическая кома
Причины:
- передозировка сахаросни-
жающими препаратами у
больных СД II
-

Причиной гипогликемической комы является абсолютная недостаточность глюкозы для обеспечения энергетических процессов
Причиной гипогликемической комы является абсолютная недостаточность глюкозы для обеспечения энергетических процессов

Хронические осложнения сахарного диабета
К хроническим осложнениям СД относятся:
ангиопатии (микроангиопатии; макроангиопатии)
полинейропатии
кардиоваскулярная патология
артериальная
Хронические осложнения сахарного диабета
К хроническим осложнениям СД относятся:
ангиопатии (микроангиопатии; макроангиопатии)
полинейропатии
кардиоваскулярная патология
артериальная

1. Неферментативное гликозилирование белков базальных
мембран капилляров, приводящее:
а) к «сшивке»
1. Неферментативное гликозилирование белков базальных
мембран капилляров, приводящее:
а) к «сшивке»

Последствия микроангиопатии
Набухание, утолщение и дистрофия эндотелия сосудов.
Изменение строения белков
Последствия микроангиопатии
Набухание, утолщение и дистрофия эндотелия сосудов.
Изменение строения белков

1 Увеличение концентрации глико- и мукопротеидов и
отложением гиалина
1 Увеличение концентрации глико- и мукопротеидов и
отложением гиалина

8 Накопление сорбитола в стенке артериальных сосудов.
9 Активация синтеза
8 Накопление сорбитола в стенке артериальных сосудов.
9 Активация синтеза

КАРДИОВАСКУЛЯРНАЯ ПАТОЛОГИЯ
является основной причиной, вызывающей высокую летальность у больных сахарным
КАРДИОВАСКУЛЯРНАЯ ПАТОЛОГИЯ
является основной причиной, вызывающей высокую летальность у больных сахарным

Патогенез нарушений деятельности сердца
- возникновение дистрофических изменений в миокарде за счет
Патогенез нарушений деятельности сердца
- возникновение дистрофических изменений в миокарде за счет

Причины:
Гиперинсулинемия способствует задержке в организме Na. При этом
Na потенцирует действие
Причины:
Гиперинсулинемия способствует задержке в организме Na. При этом
Na потенцирует действие

Основные звенья патогенеза диабетической нейропатии:
Снижение интраневрального кровоснабжения в связи с
Основные звенья патогенеза диабетической нейропатии:
Снижение интраневрального кровоснабжения в связи с

Патогенез нефропатий
Нарушение функций почек - одна из частых причин инвалидизации
и смерти
Патогенез нефропатий
Нарушение функций почек - одна из частых причин инвалидизации
и смерти

Ретинопатия
Поражение сетчатки глаза при диабете выявляют примерно у 3% больных в
Ретинопатия
Поражение сетчатки глаза при диабете выявляют примерно у 3% больных в
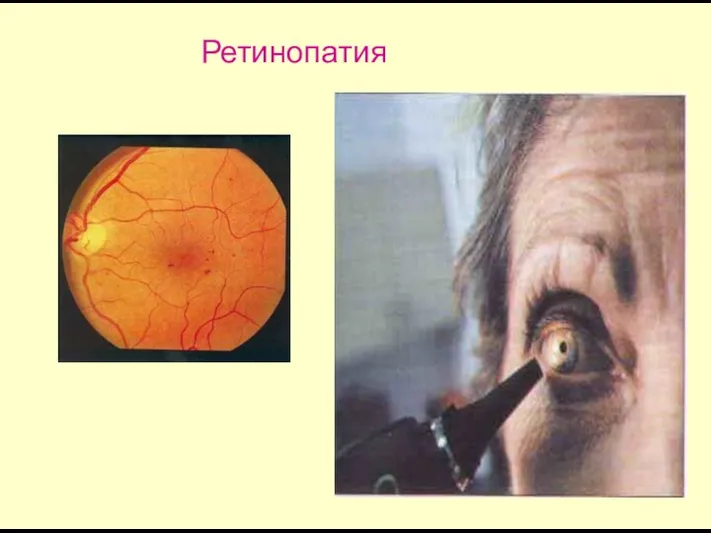
Ретинопатия
Ретинопатия
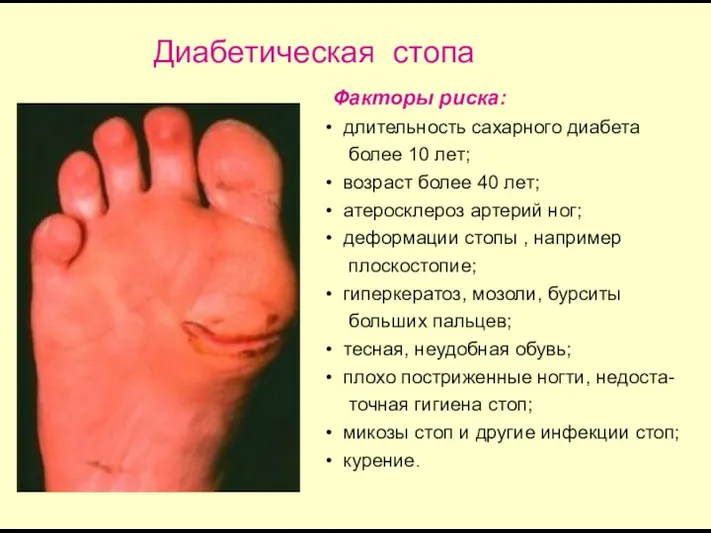
Диабетическая стопа
Факторы риска:
длительность сахарного диабета
более 10 лет;
Диабетическая стопа
Факторы риска:
длительность сахарного диабета
более 10 лет;
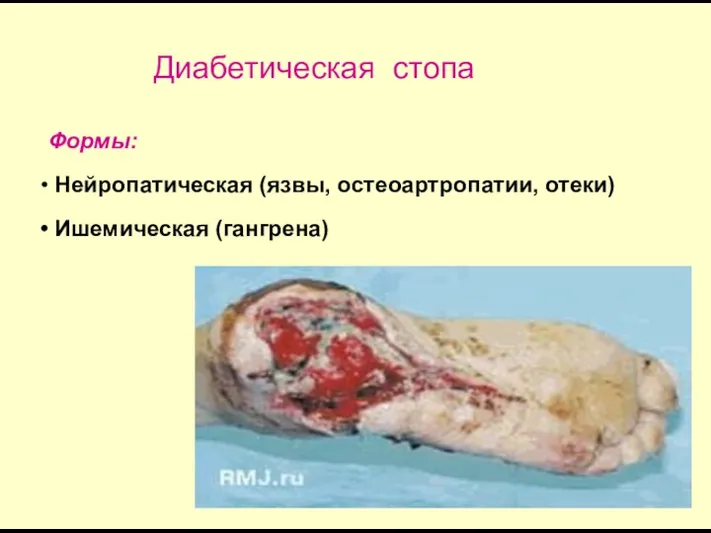
Формы:
Нейропатическая (язвы, остеоартропатии, отеки)
Ишемическая (гангрена)
Диабетическая стопа
Формы:
Нейропатическая (язвы, остеоартропатии, отеки)
Ишемическая (гангрена)
Диабетическая стопа
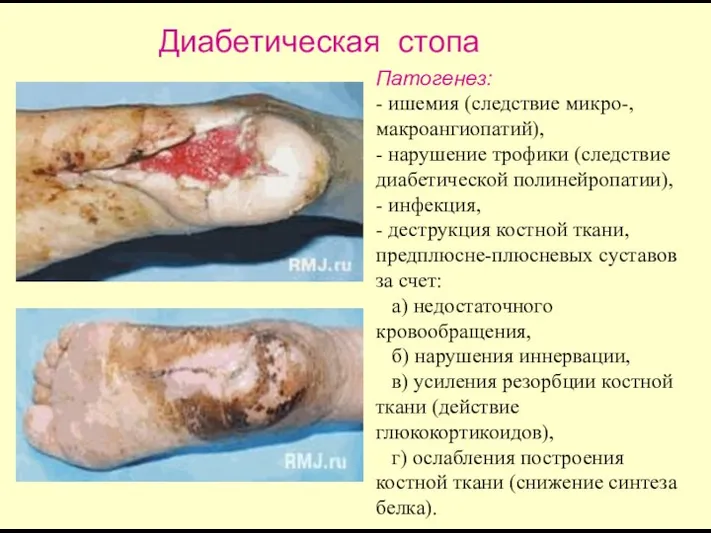
Патогенез:
- ишемия (следствие микро-, макроангиопатий),
- нарушение трофики (следствие диабетической полинейропатии),
- инфекция,
-
Патогенез:
- ишемия (следствие микро-, макроангиопатий),
- нарушение трофики (следствие диабетической полинейропатии),
- инфекция,
-
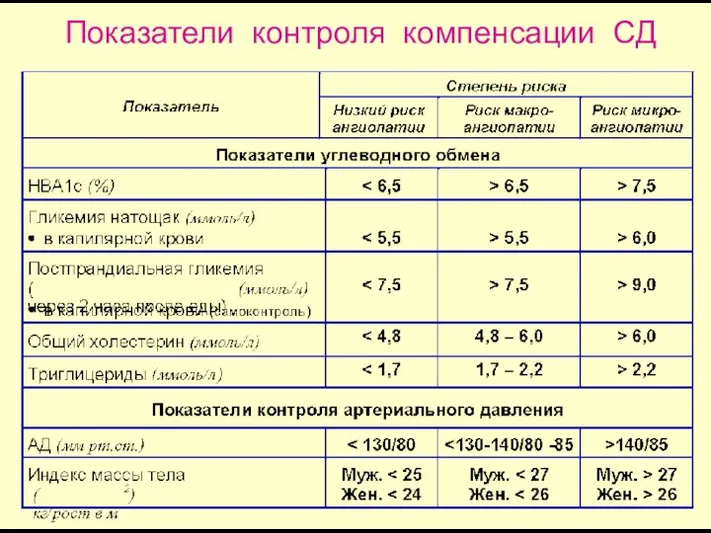
Показатели контроля компенсации СД
Показатели контроля компенсации СД

1. Этиологический – направлен на устранение причины
на начальном этапе
2. Патогенетический
1. Этиологический – направлен на устранение причины
на начальном этапе
2. Патогенетический

В терапии СД 2 типа в последние годы уделяют большое внимание
В терапии СД 2 типа в последние годы уделяют большое внимание

Воспроизведение сахарного диабета в эксперименте
Основные сведения об этиологии и патогенезе сахарного
Воспроизведение сахарного диабета в эксперименте
Основные сведения об этиологии и патогенезе сахарного

Широкое распространение получила модель аллоксанового диабета, возникающего при введении животным аллоксана.
Широкое распространение получила модель аллоксанового диабета, возникающего при введении животным аллоксана.
 Несеп – жыныс жүйесінің ауытқулары
Несеп – жыныс жүйесінің ауытқулары Адаптация пациентов к зубным протезам. Механизм и динамика адаптации
Адаптация пациентов к зубным протезам. Механизм и динамика адаптации Hypocortisolism Addison's disease
Hypocortisolism Addison's disease Студенттердің тамақтану ерекшеліктері
Студенттердің тамақтану ерекшеліктері ЛФК при переломах грудных и поясничных позвонков
ЛФК при переломах грудных и поясничных позвонков Менигококковая инфекция
Менигококковая инфекция Дифференциальная диагностика инфаркта миокарда
Дифференциальная диагностика инфаркта миокарда Основные положения гигиенической оценки условий труда в процедуре аттестации рабочих мест
Основные положения гигиенической оценки условий труда в процедуре аттестации рабочих мест Нейроинфекция у детей
Нейроинфекция у детей Тірі адамға сараптама жасау
Тірі адамға сараптама жасау An Introduction To The Health Effects of Arsenic (As)
An Introduction To The Health Effects of Arsenic (As) Консультация учителя-дефектолога: Основные направления коррекционной работы по исправлению недостатков звукопроизношения
Консультация учителя-дефектолога: Основные направления коррекционной работы по исправлению недостатков звукопроизношения Мукополисахаридоз
Мукополисахаридоз Сальмонеллез, колибактериоз, диплококкоз сельскохозяйственных животных
Сальмонеллез, колибактериоз, диплококкоз сельскохозяйственных животных Технология мягких лекарственных форм
Технология мягких лекарственных форм Эхинококкоз человека
Эхинококкоз человека Ультрадыбыстың медицинада қолданылуы
Ультрадыбыстың медицинада қолданылуы Наркомании и токсикомании
Наркомании и токсикомании Воспаление. Причинные факторы воспаления (флогогены)
Воспаление. Причинные факторы воспаления (флогогены) Удаление дивертикула Меккеля. Болезнь Гиршпрунга
Удаление дивертикула Меккеля. Болезнь Гиршпрунга Методы исследования больных с заболеваниями органов дыхания. Расспрос больного
Методы исследования больных с заболеваниями органов дыхания. Расспрос больного Септический шок и септицемия
Септический шок и септицемия Гипотермия и Гипертермия
Гипотермия и Гипертермия Портфели влияют на нашу осанку
Портфели влияют на нашу осанку Системы нижней полой вены и воротной вены. Ситуационные задачи
Системы нижней полой вены и воротной вены. Ситуационные задачи Менструальный цикл
Менструальный цикл Преэклампсия. Гестоз - осложнение беременности
Преэклампсия. Гестоз - осложнение беременности Обезболивание при стоматологическом вмешательстве у детей. Виды обезболивания. Современные анестетики, их свойства
Обезболивание при стоматологическом вмешательстве у детей. Виды обезболивания. Современные анестетики, их свойства